HOW CAN WE HELP YOU? Call 1-800-TRY-CHOP

In This Section
Search IRB Resources
Consent Form Templates
Published on Jun 15, 2022 · Last Updated 9 months 3 weeks ago
The templates on this page are intended to help investigators construct documents that are as short as possible and written in plain language. The consent form (ICF) templates provided by the IRB comply with federal regulations and HIPAA. There are other webpages devoted to providing guidance for writing readable, compliant ICFs.
Standard Language
The IRB has assembled a compendium of procedure descriptions and their associate risks from consent forms. The document can be found on the Standard Language page. To expedite approval of their consent forms, investigative teams should use these examples. The examples should serve as a starting point and should be edited as necessary to match the requirements of the specific study.
Getting Your Consent Form (ICF) Approved Quickly
To create clear, simple consent documents:
- Read and follow the IRB's advice on writing Consent Documents
- Follow the instructions in the CHOP IRB's template;
- Adhere to the template design specified in the MS Word Style Sheet - margins, type size, font choices, use of bold, etc. - which can be used to control formatting;
- Use Plain Language instead of technical terms;
- Use or adapt the descriptions of procedures and their risks that have been compiled by the CHOP IRB from approved consent forms in from the Standard Language webpage. The IRB has compiled a document entitled Explanations of Procedures and their Associated Risks which includes Plain Language descriptions of procedures and corresponding risk information. The document can be downloaded either as a pdf file or as an MS Word file. The IRB does not intend that investigators must use the exact wording; both the descriptions of the procedures and the associated risks may be modified and adapted to match each study's specific requirements;
- Target the reading level as close as possible to Grade 6 - 8;
- Use graphical representations to explain risk (low numeracy is an even bigger problem than low literacy level);
- Have someone without a medical background who is unfamiliar with the study review the consent form;
- Edit, revise and edit again - until the document is clear and concise.
What if the sponsor or study group has provided a model consent form?
Those rewriting model consent forms to fit the CHOP consent form templates often make them worse rather than better. The IRB will accept and approve an ICF that differs from the templates, provided that the document contains all of the required and any applicable optional elements and it is well written. The consent form must include the same required elements as a consent form where another IRB serves as the reviewing IRB (CHOP ICF Requirements). This includes the header should identify CHOP as the site, the CHOP PI (and not an external PI) is listed with their contact information, CHOP's HIPAA language should be used, CHOP's Injury Compensation Language must be used, the CHOP footer must be at the bottom of the ICF so that the eIRB system can stamp the form.
Tips for Developing a New Consent Form
Writing a consent form requires substantial effort to take the consent template and add the required information using plain language, brevity and clarity. However, making the effort at the time of the initial submission will greatly speed up the approval of the study. To get your consent form approved quickly, it is incumbent on the investigative staff to create clear, simple consent documents.
 The PRISM Readability Toolkit contains information about the principles of Plain Language and methods and examples for improving readability. The toolkit recommends using the Flesh-Kincaid reading tool included in Microsoft Word. However, the CHOP IRB recommends using the tools available storytoolz.com instead, particularly the SMOG index. Studies have shown that the Flesh-Kincaid can underestimate the difficulty of text related to medical information by 1 - 2 grades.
The PRISM Readability Toolkit contains information about the principles of Plain Language and methods and examples for improving readability. The toolkit recommends using the Flesh-Kincaid reading tool included in Microsoft Word. However, the CHOP IRB recommends using the tools available storytoolz.com instead, particularly the SMOG index. Studies have shown that the Flesh-Kincaid can underestimate the difficulty of text related to medical information by 1 - 2 grades.
Pictographs that present a graphical representation of the frequency of study risks, such as the meaning of likely, less likely and rare or common, occasional, and rare have been shown to improve subject comprehension. Two examples are included below so that they can be incorporated into consent documents.
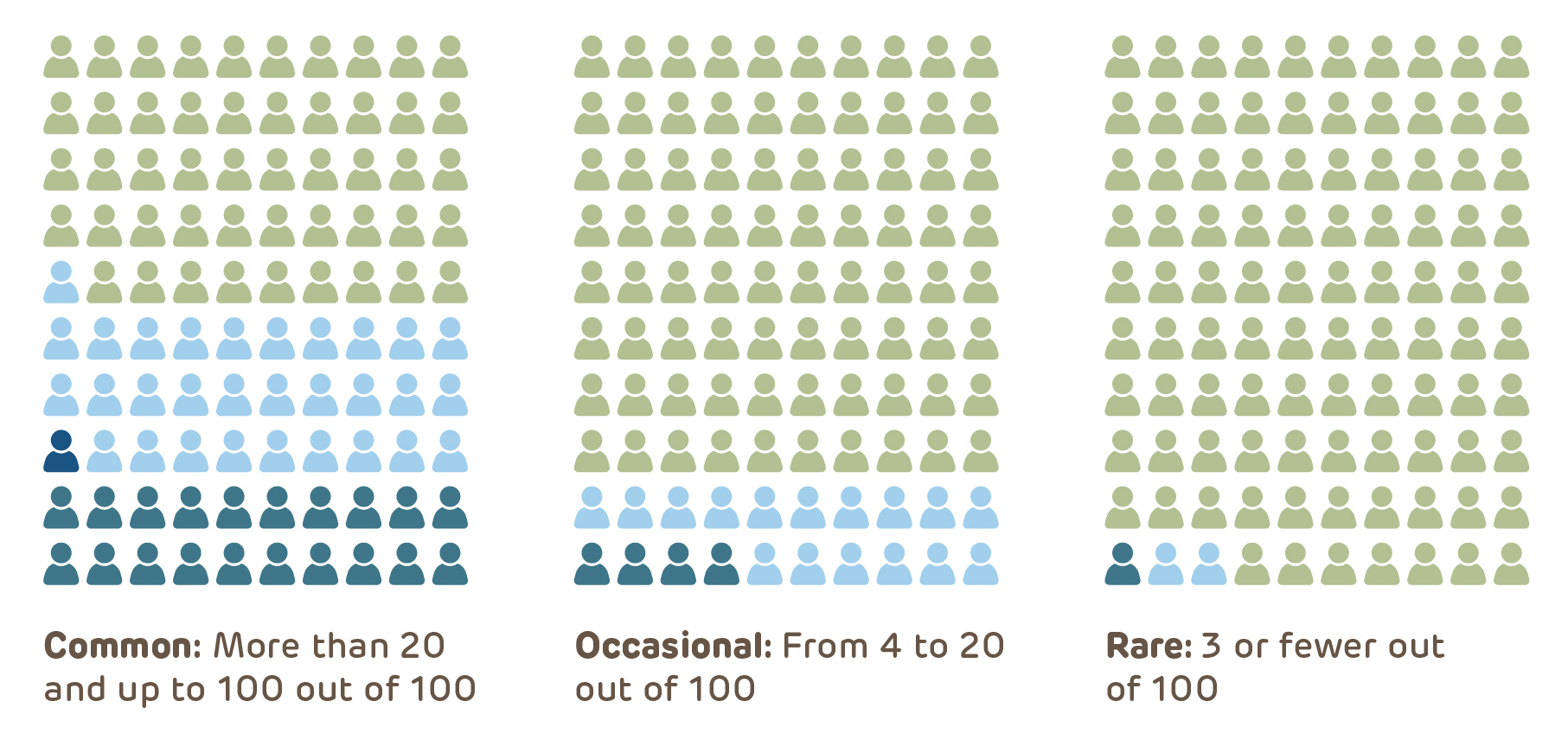
When HIPAA applies to the research, the subject must provide HIPAA authorization in addition to informed consent. This can be in the form of either
- A combined consent/HIPAA authorization which is a single document that meets the regulatory requirements for research consent and HIPAA; or
- A consent form and a stand-alone HIPAA authorization form.
The combined consent/HIPAA authorization has the advantage of requiring a single document and requires a subject's signature on only a single document. The disadvantage is that the consent form is longer and is harder to understand.
Using a stand-alone HIPAA Authorization instead of a combined document shortens and simplifies the consent document considerably. There is another advantage to using a stand-alone HIPAA Authorization, the IRB does not review or approve stand-alone HIPAA Authorizations. The responsibility for this form falls on the investigator. There are disadvantages, subjects will need to sign twice - once for the consent form and and once for the HIPAA Authorization - instead of a signing a just one document. In addition, the investigator will need to remember to use both forms instead of just a single form.
The current versions of the CHOP informed consent templates are compliant with the FDA's new required statement for clinical trials. This statement is only required when there is an FDA requirement for trial registration on clinicaltrials.gov (see below). This new requirement applies to all new and revised consent forms subject to FDA regulations and reporting requirements as of March 7, 2011.
21 CFR 50.25 - Elements of Informed Consent
(c) When seeking informed consent for applicable clinical trials, as defined in 42 U.S.C. 282(j)(1)(A), the following statement shall be provided to each clinical trial subject in informed consent documents and processes. This will notify the clinical trial subject that clinical trial information has been or will be submitted for inclusion in the clinical trial registry databank under paragraph (j) of section 402 of the Public Health Service Act. The statement is:
A description of this clinical trial will be available on http://www.ClinicalTrials.gov, as required by U.S. Law. This Web site will not include information that can identify you. At most, the Web site will include a summary of the results. You can search this Web site at any time.
Consent Templates and Other Forms
| Combined Consent Form and HIPAA Authorization Templates (in compliance with the 2018 Common Rule) | Download | Version |
|---|---|---|
| Combined Informed Consent with HIPAA Authorization This ICF template combines the required elements of consent (in accordance with the 2018 Common Rule) as well as the required HIPAA statements into a single form. For ease of navigation, the additional consent elements required by the 2018 Common Rule are indicated in green font. This version includes the Certificate of Confidentiality language, which is now required for all NIH-funded studies. |
ICF with HIPAA | 6-25-2023 |
| Combined Informed Consent with HIPAA Authorization and Study Summary Document Signature Pages Instead of creating a separate Study Summary Document to use with the Short Form Consent process, it is possible to create a single form that includes the Study Summary Document Signature pages at the end. This ICF Template includes the standard combined ICF - HIPAA template plus the final two pages include the Study Summary Document signature pages. |
ICF with HIPAA authorization and Study Summary Document signature pages | 6-25-2023 |
| Integrating 2018 Common Rule Requirements Into Currently Approved Consent Forms | Download | Version |
|---|---|---|
| Updating Consent Forms to the 2018 Common Rule Requirements This document provides guidance on how to integrate the 2018 Common Rule requirements into a currently approved consent form (or a consent form currently in review for a not yet approved study). |
ICF Update to 2018 Common Rule | 2-12-2019 |
| Concise Summary Templates(as required by the 2018 Common Rule) | Download | Version |
|---|---|---|
| Concise Summary for Greater than Minimal Risk Studies This template is an example for a study involving a study drug and can be customized for other greater than minimal risk studies. |
CS greater than minimal risk | 4-19-2019 |
| Concise Summary for Minimal Risk Studies This template can be used for studies involving no more than minimal risk. |
CS minimal risk | 4-19-2019 |
| Concise Summary for Biorepository Studies This template can be used for studies establishing a biorepository. |
CS Biorepository | 4-19-2019 |
| Consent Form (without HIPAA authorization) and Stand-Alone HIPAA Authorization (in compliance with the 2018 Common Rule) | Download | Version |
|---|---|---|
| Informed Consent Template (without HIPAA) This ICF template includes the required elements of consent without the required elements of HIPAA. It is intended for use in situations where either (1) HIPAA does not apply or (2) when a stand-alone Written Authorization will be used instead of a combined consent-HIPAA authorization form. |
ICF without HIPAA | 6-25-2023 |
| Stand-Alone Written Authorization (HIPAA) This is a CHOP-approved template for Written Authorization. The IRB provides this template, which has been reviewed by the CHOP Privacy Office as a service to investigators. |
Stand-alone HIPAA Authorization | 10-7-2016 |
| Specialized Consent Forms (in compliance with the 2018 Common Rule) | Download | Version |
|---|---|---|
| Informed Consent Template for Screening Procedures This consent form is designed specifically for obtaining either verbal or written consent to screen potential subjects in order to determine whether or not they are eligible to participate in a research study. The signature blocks are available both for documenting verbal consent (useful for subjects contacted by phone) and written consent, whichever is applicable. There are directions for creating an ICF for Verbal Consent at the end of the table. More information concerning consent and HIPAA authorization (when applicable) for screening is available on Recruitment vs Screening. |
ICF: Screening Procedures | 6-25-2023 |
| Consent Template for a Biorepository (or Registry) This consent form is an example, designed specifically for Registries and Biorepositories. It is assumed that this version will be used as a starting point and might need extensive modifications to adapt to study requirements. The example assumes that specimens are being collected for future research and that there is no intent to return results. The actual content will vary depending on the nature of the planned data and specimens, the frequency (if at all) that data will be updated, and under what circumstances (if at all) results could be returned to participants, and other related issues specific to the particular repository design. |
ICF for a Registry or Biorepository | 6-11-2019 |
| Consent Template for Single Patient Treatment IND or IDE (expanded access) This consent form is an example, designed specifically for Single Patient Expanded Access or Emergency Use. It is assumed that this template will be used as a starting point and might need modifications to adapt to the single patient to be treated. The example assumes the use of a drug. The actual content will vary depending on the nature of the investigational agent or device and whether procedures are done as part of the clinical investigation or clinical care. |
ICF for single patient IND or IDE | 6-25-2023 |
| Informed Consent Template for Pregnant Partner This ICF is intended for situations where a sponsor wants to follow the outcome of the inadvertent pregnancy of the partner of a study subject. Usually this includes follow up of the child after birth to ensure that there were no birth defects. |
ICF: Pregnant Partner | 6-25-2023 |
| Consent Template Exempt Research This consent form is an example, designed specifically for Exempt survey research and is provided purely as a service by the IRB. The IRB does not review or approve the content of exempt consent forms. The consent form should not include any mention of IRB approval and it should not include the standard IRB footer (the eIRB system does not stamp exempt consent forms). This consent form should be edited and revised to match the needs of the research. |
Exempt Study Consent Template | 6-25-2023 |
| Optional Sections for the Consent Form and Consent Addenda | Download | Version |
|---|---|---|
| Consent Addendum This is a consent addendum to allow already enrolled participants to agree to additional study procedures not disclosed in the initial consent form. This form supplements the consent and HIPAA authorization the subject already provided for a research study. |
Consent Addendum | 3-11-2020 |
| Optional Section for Sharing Data with the NIH This section should be included in the consent form when there is an intent to share data with the NIH data registry. While sharing can be restricted or unrestricted, it is easier to track if limited to unrestricted sharing. |
Sharing Data with the NIH | 3-21-2018 |
| CHOP-approved Certificate of Confidentiality (CoC) This is an MS Word document with the the CoC Language approved by the CHOP IRB and CHOP legal counsel. |
CHOP CoC Text | 3-8-2018 |
| Return of Results (RoR) Consent Addendum for Genetic Research This is an addendum to be used in conjunction with the consent form to allow participants to choose which genetic research results they would like to have returned to them. |
RoR Addendum | 6-13-2019 |
The consent form templates include a signature page that is appropriate for studies approved under 45 CFR 46.404 or 405. There are many studies that involve circumstances that require revision to the standard language, for example, studies that include both parents as subjects or only adults. The signature pages below can be used to substitute for the standard signature page for studies where the participants differ from the most common scenarios.
| Signature Pages | Download | Version |
|---|---|---|
| Standard Signature Page: Child and Adult Participants This is the standard signature page included in the IRB consent form templates. It is appropriate for use for studies approved by the IRB under 45 CFR 46.404 or 405. It can be used when both children and adults are study participants and can be adapted to include one of the child's parents as a participant. |
Standard Child or Adult | 3-11-2015 |
| Alternate Child-Adult Signature Page This an alternate version that separates the consent text for the adult participants from the child (or child and parent) participants. |
Alternate Child or Adult | 5-15-2014 |
| Signature Page for a Child and Both Parents This version contains the standard language and also includes signature blocks for both parents to sign when they are also subjects of the research |
Child + Both Parents | 5-15-2014 |
| Signature Page for Adult Participants This signature page should be used when the only participants are adults or minors that may consent for themselves. |
Adult Participant | 5-15-2014 |
| Signature Page for Adult, Adult with Diminished Capacity and Child Participants This signature page should be used when the participants may include adults, adults with diminished capacity and children. |
Adult, Adult with Diminished Capacity or Child Participant | 10-16-2015 |
| Signature Page Study Summary Document for Adult, Adult with Diminished Capacity and Child Participants This signature page should be used study summary documents when the participants may include adults, adults with diminished capacity and children. |
Adult, Adult with Diminished Capacity or Child Participant Study Summary Document | 10-16-2015 |
| Signature Page for 406 Study - Child Participants Studies approved under 45 CFR 46.406 require the signatures of two parents. This version should be used when the participants are limited to those less than 18 years of age. |
406 Child Participant | 5-15-2014 |
| Signature Page for 406 Study - Child and Adult Participants These signature blocks should be used when either children or adults may participate and the study is approved by the IRB under 45 CFR 46.406 (signature lines for both parents). |
406 Child or Adult Participant | 5-15-2014 |
What is commonly known as verbal consent is in regulatory terms referred to as informed consent with waiver of documentation. The same elements of informed consent and HIPAA authorization (if applicable) are required. The IRB does not recommend using a "verbal script"; we recommend that the investigators develop a consent form to that will be read to the prospective subject. One of the standard consent templates can be modified for this purpose as follows:
- Create a consent form using one of the standard consent form templates (above);
- Replace the standard signature pages with one of the two Documentation of Consent Signature pages (below);
- The person obtaining consent signs and dates the consent form documenting that the consent discussion took place.
- If consent will be documented in the study record or by some other method, it is not necessary to include the Documentation page.
| Verbal Consent Signature Pages | Download | Version |
|---|---|---|
| Verbal Consent Form: Documentation of Consent Signature Page This page may be used to replace the standard signature page when the IRB has waived the requirement for documentation of consent. This process is referred to colloquially as verbal consent. |
(Verbal) Consent Signature Page | 5-12-2014 |
| Verbal Consent Form: Documentation of Consent and Assent Signature Pages These pages may be used to replace the standard signature pages when the IRB has waived the requirement for documentation of consent and assent. This process is referred to colloquially as verbal consent. |
(Verbal) Consent and Assent Signature Pages | 3-11-2015 |
When using a Short Form Consent process for non-English speaking subjects, the Study Summary document replaces the standard consent form. There are two differences: (1) title changes to "Study Summary Document" and (2) there is a different signature page which is signed by the member of the investigative team obtaining consent and the witness instead of the subject (or parent of the subject).
| Signature Pages for Study Summary Documents | Download | Version |
|---|---|---|
| Summary Document Signature Page The signature block may be modified to meet the circumstances appropriate to the study. These pages may be used with a combined consent/study summary document or with a stand-alone study summary document. |
Summary Document Signature Blocks | 3-11-2015 |
| Summary Document Signature Page for 406 Study Studies approved under §46.406/50.53 require two parents signatures. The signature block may be modified to meet the circumstances appropriate to the study. This page may be used either with a combined consent/study summary document or with a stand-alone study summary document. |
Summary Document Signature Blocks for §406 Study | 8-29-2016 |