HOW CAN WE HELP YOU? Call 1-800-TRY-CHOP

In This Section
Search IRB Resources
Emergency Use
Published on Jun 16, 2022 · Last Updated 1 month 1 week ago
The need for an investigational drug or biologic may arise in an emergency situation that does not allow time for submission of an IND. The FDA can authorize shipment of the test article (drug, biologic or device) in advance of the IND submission. Requests for an authorization may be made by telephone, fax or email using the FDA contact information provided in 21 CFR 312.310 or the FDA Information Sheets.
The FDA requirements and regulations for an emergency IND for the use of an investigational drug or biologic are slightly different than for an emergency use of a device. In both situations, FDA must conclude that the use is for a "serious or life-threatening disease or condition and there is no comparable or satisfactory alternative therapy to diagnose, monitor, or treat the disease or condition."
Emergency Use is a special case of the Expanded Access to Investigational Drugs for Treatment Use. Most of the time, the IRB will have sufficient time to review investigator's requests for expanded access to an investigational drug. Even though only a single individual (or a small group) will receive the investigational drug, this is considered research (a clinical investigation) under the FDA regulations.
2017 FDA Guidance: IRB Concurrence
On October 3, 2017, the FDA issued new guidance regarding individual patient expanded access INDs. The new pathway still involves a submission to the IRB, but the IRB can now provide concurrence by the chair or another IRB member (rather than review and approval by the convened board). To use this pathway, the sponsor-investigator needs to request 'Authorization to Use Alternative IRB Review Procedures' from the FDA. This can be done either on form FDA 3926 (section 10.b.), or, if submitting using a 1571, by a separate attachment to the FDA. Note: This pathway is only available when a physician sponsor-investigator will hold the IND; it is not available when the pharmaceutical sponsor will hold the IND.
To obtain concurrence, the sponsor-investigator should select "Clinical Investigation involving a single human subject (e.g. emergency IND or IDE); CHOP IRB has time to review" in eIRB, complete the abbreviated submission and attach the documentation submitted to the FDA (e.g. Form 3926). For a single patient treatment IND consent template, go to Consent Template for Single Patient Treatment IND or IDE (expanded access). For assistance with single patient IND submissions to the FDA, contact CHOP’s IND/IDE Support Program.
FDA Resources for Expanded Access and Emergency Use
FDA 24/7 Contact Information:
The FDA mans a hotline 24 hours a day, 7 days a week for emergency requests. Click on the links for the FDA contact information.
- Physician Request for an Individual Patient IND under Expanded Access for Non-emergency or Emergency Use or
- FDA's Expanded Access Contact Information.
Guidance and Forms for Expanded Access to Investigational Drugs for Treatment Use
- FDA's Guidance - Expanded Access to Investigational Drugs for Treatment Use - Questions and Answers covers issues related to the treatment use of investigational drugs for a single individual, as well as intermediate and large size trials.
- Form FDA 3926 – Individual Patient Expanded Access Investigational New Drug Application (IND)
- Instructions for Filling Out Form 3926: Individual Patient Expanded Access IND
FDA Websites with Educational Materials about Expanded Access to Drugs and Devices:
- FDA page for Physician Request for an Individual Patient IND under Expanded Access for Non-emergency or Emergency Use.
- FDA page with educational information regarding IDE Early/Expanded Access.
Single Subject IND: Emergency Use of a Drug or Biologic
Definitions
21 CFR 312.300(b)?
Immediately life-threatening disease or condition:
A stage of disease in which there is reasonable likelihood that death will occur within a matter of months or in which premature death is likely without early treatment.
Serious disease or condition:
A disease or condition associated with morbidity that has substantial impact on day-to-day functioning. Short-lived and self-limiting morbidity will usually not be sufficient, but the morbidity need not be irreversible, provided it is persistent or recurrent. Whether a disease or condition is serious is a matter of clinical judgment, based on its impact on such factors as survival, day-to-day functioning, or the likelihood that the disease, if left untreated, will progress from a less severe condition to a more serious one.
21 CFR 56.102(d)
Emergency use:
The use of a test article on a human subject in a life-threatening situation in which no standard acceptable treatment is available, and in which there is not sufficient time to obtain IRB approval.
General Requirements:
The requirements for emergency use of a drug are included in the new (2009) 21 CFR 312 Subpart I: Expanded Access to Investigational Drugs for Treatment Use. Expanded Access includes single patient use, intermediate-size population use, and treatment use or treatment INDs. The requirements for all three types of expanded access in 21 CFR 312.305.
Specific Requirements for Single Patient IND:
The detailed requirements for a single patient use, including emergency use, can be found in 21 CFR 312.310.
IRB Requirements:
For drugs and biologics, the FDA regulations do not provide for expedited IRB approval in emergency situations. The FDA therefore, does not accept any of the following, though commonly used terms - "interim," "compassionate," or "temporary". There are three possible ways to proceed:
-
the IRB can provide concurrence by the chair or another IRB member for the emergency use (see above), provided the FDA has granted a waiver under § 56.105 of the requirements in § 56.108(c), which relate to IRB review by the convened board;
-
the IRB must convene and give full board approval for the emergency use (e.g. if the sponsor is a pharmaceutical company and not a physician sponsor-investigator, or the FDA has concluded that a waiver of the requirement for review and approval by a convened board is not appropriate); or,
-
if the conditions of 21 CFR 56.102(d) are met (see information box to the right) and it is not possible to convene a quorum/chair concurrence within the time available, the use may proceed without any IRB approval/chair concurrence. This latter option is also known as an Emergency Exemption from prospective IRB review.
Subsequent Uses of an Investigational Drug or Biologic
The Emergency Exemption is for a single patient use. If the investigator anticipates the need to use the same drug for a second individual, then they must prepare a protocol for IRB approval for the proposed use.
Emergency Use of an Investigation Drug or Biologic - Information Sheet:
"The exemption, which may not be used unless all of the conditions described in 21 CFR 56.102(d) exist, allows for one emergency use of a test article without prospective IRB review. FDA regulations require that any subsequent use of the investigational product at the institution have *prospective IRB review and approval. FDA acknowledges, however, that it would be inappropriate to deny emergency treatment to a second individual if the only obstacle is that the IRB has not had sufficient time to convene a meeting to review the issue."
Emergency Exemption from Prospective IRB Review: Drugs and Biologics
Immediate life-threatening situations typically occur with short notice although occasionally the planned use can be foreseen 3 or 4 weeks in advance. When there is sufficient time, the IRB must review the proposed single patient use. When there is insufficient time for IRB review, the Investigator may exercise the Emergency Exemption from prior IRB review and administer the investigational drug.
Investigator Responsibilities When Exercising the Emergency Exemption
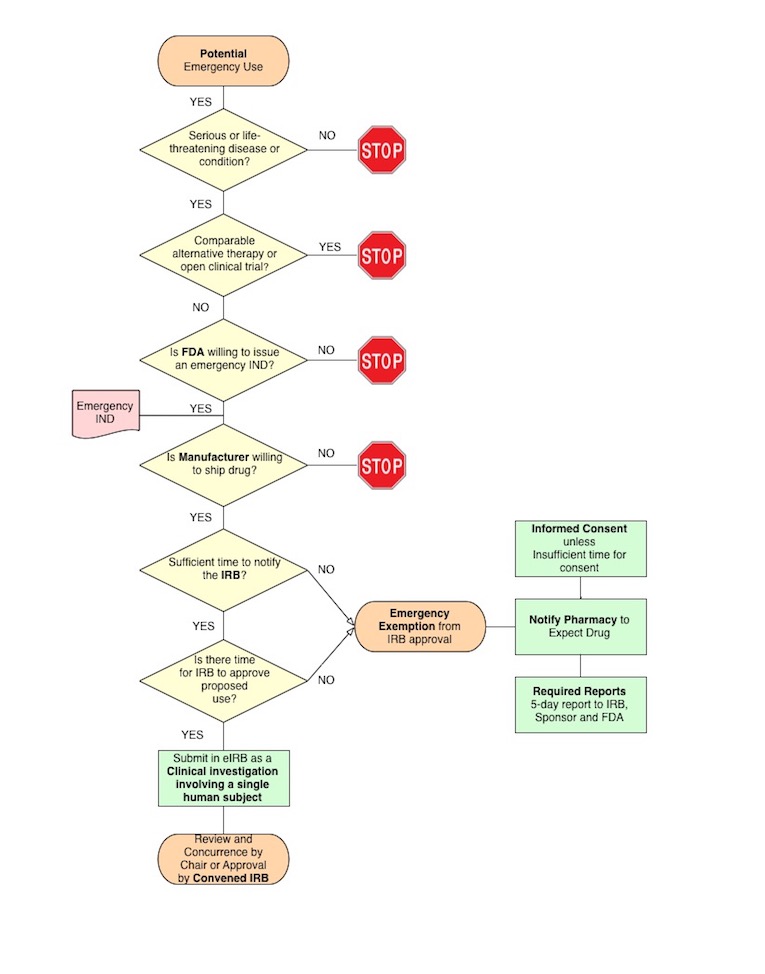
Contact the Manufacturer:
The investigator must obtain the manufacturer's agreement to ship the drug or to use part of the drug supply available at CHOP as part of another clinical trial.
Contact the FDA:
Either the investigator or the sponsor-IND holder must obtain an IND from the FDA for the emergency use. The FDA maintains 24 hour coverage for emergency INDs (see above).
Contact the IRB:
When there is sufficient time, the investigator should contact the IRB as soon as possible to determine whether or not the IRB can issue concurrence or convene and approve the emergency use. When there is insufficient time, the investigator may exercise the Emergency Exemption from prospective IRB review and must follow the procedures outlined in IRB SOP 802.
Notify the Pharmacy:
The investigator should provide the pharmacy with the following:
- a copy of the FDA's letter or email indicating its approval for the Emergency IND; the manufacturer's approval to ship drug;
- either evidence of IRB approval/concurrence or notice that the investigator is exercising the Emergency Exemption.
The drug must be shipped to the Pharmacy to maintain the appropriate chain of custody. The Pharmacy will not accept medications shipped to the investigator without proper shipping and storage information.
Report to the IRB, the FDA and Manufacturer:
If the Emergency Exemption is exercised, the IRB must be notified via the Emergency Use pathway in eIRB (check "Emergency Use of Investigational Agent" in section 1.01). There is an option to notify the IRB prior to the use. Regardless of whether an initial notification is file the investigator must complete a 5-day Report so that the IRB can ascertain that the use was appropriate. The investigator must report the use, including adverse events to the FDA.
Emergency Use of Investigational Devices
The FDA has not objected if a physician chooses to use an unapproved device in an emergency, provided that the physician later justifies that an emergency actually existed. Each of the following conditions must exist to justify emergency use of an investigational device:
- the patient is in a life-threatening condition that needs immediate treatment;
- no generally acceptable alternative for treating the patient is available; and
- because of the immediate need to use the device, there is no time to use existing procedures to get FDA approval for the use.
The FDA expects that the investigator will follow as many patient protection procedures as possible. These protection procedures include:
- Informed consent from the patient or a legal representative;
- Notify and obtain the concurrence of the IRB chairperson;
- Clearance from the institution as specified by their policies (at CHOP, this requirement is met by notification of the IRB chair or vice-chair or the AVP, Research Compliance and Regulatory Affairs);
- An independent assessment by an uninvolved physician; and
- Authorization from the IDE holder, if an approved IDE for the device exists.
The investigator should do the following:
- Report to the IRB within five days and otherwise comply with provisions of the IRB regulations (21 CFR part 56);
- Evaluate the likelihood of a similar need for the device occurring again, and if future use is likely, immediately initiate efforts to obtain IRB approval and an approved IDE for the device's subsequent use; and
- If an IDE for the use does exist, notify the sponsor of the emergency use, or if an IDE does not exist, notify FDA of the emergency use and provide FDA with a written summary of the conditions constituting the emergency, subject protection measures, and results.
Subsequent emergency use of the device may not occur unless the physician or another person obtains approval of an IDE for the device and its use.
FDA's Expectations for Investigator-Sponsors Holding a Single Subject IND or IDE
When an investigator holds the IND or IDE for a single subject, they are none-the-less the study sponsor. The FDA's enumerates the responsibilities of sponsors at 21 CFR 312.50 - 70 (Subpart D) Responsibilities of Sponsors and Investigators. An example of the type of letter that the FDA will send confirming that they have granted the single-subject IND are included below.


Exception From Informed Consent Requirement
Even for an emergency use, the investigator is required to obtain informed consent of the subject or the subject's legally authorized representative unless both the investigator and a physician who is not otherwise participating in the clinical investigation certify in writing all of the following 21 CFR 50.23 Exceptions from the general requirements:
- The subject is confronted by a life-threatening situation necessitating the use of the test article.
- Informed consent cannot be obtained because of an inability to communicate with, or obtain legally effective consent from, the subject.
- Time is not sufficient to obtain consent from the subject's legal representative.
- No alternative method of approved or generally recognized therapy is available that provides an equal or greater likelihood of saving the subject's life.
If, in the investigator's opinion, immediate use of the test article is required to preserve the subject's life, and if time is not sufficient to obtain an independent physician's determination that the four conditions above apply, the clinical investigator should make the determination and, within 5 working days after the use of the article, have the determination reviewed and evaluated in writing by a physician who is not participating in the clinical investigation. The investigator must notify the IRB within 5 working days after the use of the test article (21 CFR 50.23(c)).
CHOP Clinical Research Support Office: IND/IDE Support Program
Gregory Podsakoff, M.D.
Director, Clinical Trials Research
Phone: 267-426-5400
Email: indide [at] chop.edu (indide[at]chop[dot]edu)
Website: CHOP’s IND/IDE Support Program
US Food and Drug Administration
- 21 CFR 312: Subpart I This subpart includes the FDA regulations deal for all aspects of expanded access to investigational drugs, including emergency use.
- The Food and Drug Administration Perspective: Use of an Investigational Drug in a Medical Emergency
- Expanded Access to Investigational Drugs for Treatment Use — Qs & As. Draft Guidance
- 21 CFR 812.36 Treatment use of an investigational device
- IDE Guidance
- IDE Early/Expanded Access
- Emergency and Compassionate Use of Unapproved Devices (lecture presented by Dr. F. Santel at CDRH, FDA)