HOW CAN WE HELP YOU? Call 1-800-TRY-CHOP

In This Section
Search IRB Resources
What do I need to know about consent, and how do I write a consent form?
Published on Jun 20, 2022 · Last Updated 1 year 6 months ago
Writing Readable Informed Consent Forms (ICFs)
45 CFR 46.116
Regulatory Requirements for Informed Consent
§46.116...The information that is given to the subject or the representative shall be in language understandable to the subject or the representative. No informed consent, whether oral or written, may include any exculpatory language through which the subject or the representative is made to waive or appear to waive any of the subject's legal rights, or releases or appears to release the investigator, the sponsor, the institution or its agents from liability for negligence.
§46.111(5) Informed consent will be appropriately documented, or appropriately waived in accordance with, §46.117.
See the section on Regulatory Requirements.
Informed consent is a process and not simply a document. The ICF informs prospective participants of the purpose, procedures and risks and benefits of participation, it serves as a guide during discussions with the investigator and as a reference source during participation.
This section provides guidance for investigators preparing consent documents. The IRB provides template consent documents but any format that is both readable and understandable is acceptable. For example, if the sponsor provides a template consent form, it can be used as the basis for the CHOP informed consent form (ICF). However, the IRB cannot approve a consent document that is not written in readable, understandable language or formatted in an approachable style.
What Needs To Be in the Consent Form?
The Common Rule and the FDA have different required and additional elements of consent. The Common Rule requirements 45 CFR 46.116 are listed above. The FDA's requirements are available in 21 CFR 50.25. The IRB checks to see if the required and applicable optional elements are present in the submitted consent form.
Language Understandable to Subjects
HHS regulations at 45 CFR 16.116 and the FDA regulations 21 CFR 50.20 also require that the consent information be in language understandable to the subject or their legally authorized representative. This means the document must be written in plain language, using lay terms, and in a language the subject understands. Language the subject understands is interpreted to mean at a level compatible with their reading comprehension.
The following sections outline the most common problems that result in requested changes.
Length and Reading Level of Consent Forms
ICFs are often too long, use medical terminology rather than lay language, and are written at too high a grade level. Investigators should aim for a 6th-8th grade reading level. Many readers cannot maintain their concentration or comprehension when an ICF exceeds 6 pages.
Even when written at a 6th grade level 20 - 30% of prospective subjects will be unable to read the document.
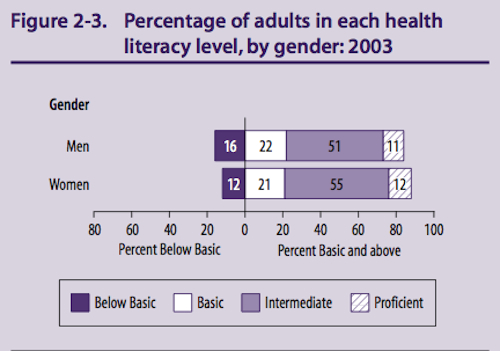
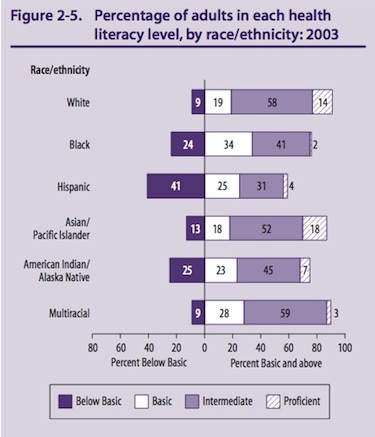
The two figures below are from the 2003 Health Literacy Statistics.
The FDA's 2014 Draft Guidance: Informed Consent Information Sheet discusses what language understandable to the subject means and points out the US adults are also severely challenged by low levels of numeracy.
“Understandable” means the information presented to potential subjects is in a language and at a level the subjects can comprehend (including an explanation of scientific and medical terms). In ensuring that information is understandable, it should be noted that more than one-third of U.S. adults, 77 million people, have basic or below basic health literacy. Limited health literacy affects adults in all racial and ethnic groups. In addition, more than one-half of U.S. adults have basic or below basic quantitative literacy and are challenged by numerical presentations of health, risk, and benefit data.
Formatting of Consent Forms
The formatting, design and layout of the ICF has been shown to be one of the most important factors that affecting readability. (See for example Tait et al. Informing the uninformed: optimizing the consent message using a fractional factorial design.) Small type, wide margins, lack of white space and inconsistent typeface usage can all make the ICF less readable. A consistent approach to the layout and design of the ICF helps the reader understand the organization of the information and this contributes to understanding.
Applying Styles in Microsoft Word Documents
Cutting and Pasting:
The IRB's informed consent form (ICF) and protocol templates contain customized styles to ensure consistent formatting. In the case of the protocol templates, they also ensure that the sections are numbered appropriately. To do this, the templates contain a collection of styles, each of which is pre-formatted for different parts of the document. Each part of the ICF and protocol template have a corresponding style which includes the font, the margins and the line spacing. For example, Heading 2 in the ICF template is Arial 12 point bold. Instead of formatting a section of text, (e.g., making text bold, or adjusting the line spacing), the correct style intended for that section of the document should be applied.
Cutting and pasting from another document will result in numerous paragraph styles, with a mixture of text sizes, and paragraph spacings. This happens because when text is pasted it brings with it the formatting from the original document intact. To prevent this problem, cut and paste using the Paste Special command and choose the Unformatted text option.
For additional information about using Style Sheets and links to Microsoft tutorials, see Preparation for Study Submission page.
A number of formatting techniques have been shown to enhance readability. All of these have been built in to the CHOP Consent Form Templates.
See the Example Informed Consent Form for a practical application of the formatting techniques described below.
- The question and answer style has been shown to result in shorter more readable ICFs;
- Keep the column width to no more than 6-6.5 inches (1.5 inch margins). Columns that are too wide are hard to read;
- Use a 12 point or larger serif typeface such as Times, Palatino, Garamond) for body text;
- Use of a sans serif typeface such as Helvetica or Arial for headings;
- Avoid the use of all UPPERCASE, it is hard to read;
- Use bold, italic, or bold italic for emphasis; minimize the use of underlining;
- Use bullet points with short phrases or sentences rather than long paragraphs for explanations;
- Double space or add additional spacing (referred to as leading) between paragraphs;
- Write in the second person, using 'you' throughout. Using the first person has been shown to be coercive.
- When children are subjects of the research, and most will be capable of assenting to participation, use 'you' to refer to the child.
- When most or all of the subjects will not be capable of providing assent (e.g. because they are under the age of 7, have cognitive deficits, will be sedated, etc.), you may use 'you' to refer to the parent or guardian and 'your child' to refer to the child.
- Be sure to edit carefully to avoid unnecessary repetition and poorly formatted documents when cutting and pasting from a sponsor's ICF template into the CHOP template.
Resources to Assist with Consent
Information on concise summaries, basic and additional elements of consent.
Template language for explanations of common study procedures and their associated risks.
Guidance on the process of obtaining written consent/parental permission.
When a subject signature is not required (e.g. to obtain verbal consent).
Guidance on when consent is not required or elements of consent can be waived.
May be permitted when it is not a reasonable requirement to protect subjects.
Use of short form consent process, when few limited English proficiency speakers are expected.
Guidance on assessing capacity and who can serve as a legally authorized representative.
Who is a child (state law) and when may minors consent for their research participation.
Consent Documents for Limited English Proficiency (LEP) Subjects
To meet the regulatory requirements for approval, the IRB must find that the selection of subjects is equitable. The investigator must either (1) justify why they are excluding LEP subjects or (2) develop a plan to allow their enrollment. For more information on translated consent forms, see Equitable Selection, Short Form Consent, and IRB SOP 701: Required Elements of Consent and Documentation of Consent on our Policies page.
-
The Group Health Cooperative in Seattle has assembled training materials for writing in plain language. Their Plain Language Toolkit is an 81 page document with numerous before and after examples. They also provide PRISM Online Training, which is an hour-long, Web-based plain language tutorial created for research professionals, including scientists, research staff, Institutional Review Boards (IRBs), or communications staff. It covers health literacy and readability, plain language strategies and examples, and interactive editing examples and exercises. Access the course for free at http://prism.grouphealthresearch.org.
-
See the Summary of the NCI's guidance related to the Checklist to Develop Easy-to-Read Consent Documents
-
Download the Glossary of Lay Terms for lay language replacements for technical terms and jargon.
-
Readability tools are available for free at - http://www.storytoolz.com/readability. The best estimate of the reading difficulty of consent forms and other health-related content is the SMOG scale.
-
MS Word can calculate readability statistics using the Flesch Reading Ease Score.The Flesch-Kincaid Grade Level score rates text on a U.S. grade-school level. Unfortunately, this scale underestimates the reading level of health-related text by one to two grade levels. If the Flesch-Kincaid is used, it is safest to add 2 grade levels. The preferred tool to use for assessing the readability of health-related information is the SMOG (Simple Measure of Gobbledygook).
-
NIH Plain Language Initiative available at - http://execsec.od.nih.gov/plainlang/intro.html
-
Plain Language available at - http://www.plainlanguage.gov
- Bjorn et al. Can the written information to research subjects be improved?--an empirical study.
- Coyne et al. Randomized, controlled trial of an easy-to-read informed consent statement for clinical trial participation: a study of the Eastern Cooperative Oncology Group.
- Franck et al. Research participant information sheets are difficult to read
- Goldstein et al. Consent form readability in university-sponsored research
- Ogloff et al. Are research participants truly informed? Readability of informed consent forms used in research
- Philipson et al. Informed consent for research: a study to evaluate readability and processability to effect change
- Shalowitz et al. Informed consent for research and authorization under the Health Insurance Portability and Accountability Act Privacy Rule: an integrated approach
- Sharp. Consent documents for oncology trials: does anybody read these things?
- Tait et al. Improving the readability and processability of a pediatric informed consent document: effects on parents' understanding
- Tait et al. Informing the uninformed: optimizing the consent message using a fractional factorial design
Frequently Asked Questions
-
What is the difference between a waiver of documentation of consent and a waiver of consent?
When the IRB grants a waiver of consent, consent is not necessary for enrollment.
When the IRB grants a waiver of documentation of consent, the investigator needs to obtain the subject’s consent but not the subject’s signature to document it. In this case, the investigator can document having obtained consent (e.g. on the verbal consent form or in the study chart).
-
Can I record any individually identifiable private information and obtain a waiver of documentation of consent under 46.117(c)(1)(i)?
In order to waive documentation under (c)(1)(i), all of the data collected must be anonymous which means recorded without any identifying information.
-
If the study is FDA-regulated, can I obtain a waiver of documentation under 46.117(c)(1)(i)?
The FDA regulations mandate that all subjects be identifiable in order to permit an audit of the source documents so there is no FDA equivalent to 46.117(c)(1)(i).
-
Which research procedures can take place and still waive documentation of consent?
To waive the requirement for documentation under 46.117(c)(1)(ii) or 56.109(c)(1), the procedures in the research must be limited to those that don't require written consent as part of clinical care. Examples include: Blood draw, Questionnaires, Chest X-ray, and DXA scan.
-
Why does the IRB require that I have a consent form approved if I have obtained a waiver of documentation of consent?
The IRB must ensure that the subject is provided with the necessary information to make an informed decision about study participation. The waiver of documentation is merely related to the requirement for a subject to sign, and thus document, their consent.
-
When do I need to give the subject an information sheet?
When the IRB waives documentation of consent under (c)(1) or under 50.109(c) of the FDA regulations, it can require the investigator to provide the subject an Information Sheet when the IRB decides that subjects should have some information to refer back to after completion of the study. The contents of the Information Sheet do not need to match those of a consent form but should contain at a minimum:
- The title of the research
- Contact information for the investigator
- An explanation of the purpose of the research
- A description of the procedures
-
What if HIPAA applies to the research, can I still obtain a waiver of documentation of consent?
HIPAA at 45 CFR 164.512 permits oral authorization instead of written authorization provided that the study meets the criteria for alteration or waiver of HIPAA. If the research involves individually identifiable health information, then the investigator must also do one of the following:
- Request an alteration (sometimes referred to as a partial waiver) of Written Authorization, or
- Obtain Written Authorization for use of PHI using a stand-alone HIPAA Authorization
-
Do I need to submit the Short Form Consent documents?
No. The IRB no longer requires the investigator to upload the short form consent documents that are already available on the IRB's website. These documents are locked and are already certified translations. The investigator only needs to submit a Study Summary Document.
-
When may the short form consent process be used instead of a translated consent form?
The short form written consent is intended when few subjects with limited English proficiency (if any) are expected to enroll.
The short form consent process is ideal for situations where a subject whose preferred language is not English presents unexpectedly and a translated consent form is not available. If many subjects with limited English proficiency are expected to enroll, then a translated consent form should be used rather than the using a short form consent document.
-
Who needs to sign the short form consent document and the summary document?
These forms are signed by the individuals who understand the information on the individual form. The short form (in the subject’s preferred language) will be signed by the subject and the interpreter/witness. The study summary document (in English) will be signed by the study team member and the interpreter/witness. The Summary Document signatures attest that the information in the document was presented to the subject and that their questions were answered. The signatures on the short form document that the subject understood the information presented.
-
Is the witness certifying the medical accuracy of the information provided by the investigator or the Study Summary Document?
No, the witness is not certifying the accuracy of the medical information. The witness is attesting that the information has been presented in a language understandable to the subject, and that the subject understood the information.