HOW CAN WE HELP YOU? Call 1-800-TRY-CHOP

In This Section
Investigational Drugs, Biologics
Published on Jun 14, 2022 · Last Updated 1 year 6 months ago
Definitions
What is a drug?
The Federal Food Drug and Cosmetic Act (FD&C Act) and the FDA regulations define the term drug, in part, by reference to its intended use, as “articles intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease” and “articles (other than food) intended to affect the structure or any function of the body of man or other animals.” Therefore, almost any ingested or topical or injectable product that, through its label or labeling (including internet websites, promotional pamphlets, and other marketing material), is claimed to be beneficial for such uses will be regulated by FDA as a drug. The definition also includes components of drugs, such as active pharmaceutical ingredients. See FDA Guidance on Human Drugs. Biological products may also be considered drugs within the meaning of the FD&C Act. Biological products (42 U.S.C. 262(i)) include, among other products, bacterial vaccines, allergenic extracts, gene therapy products, growth factors, cytokines, and monoclonal antibodies.
What is an investigational drug?
Investigational new drug means a new drug or biological drug that is used in a clinical investigation. The term also includes a biological product that is used in vitro for diagnostic purposes. The terms "investigational drug" and "investigational new drug" are deemed to be synonymous for purposes of this part. (21 CFR 312.3(b))
What is a clinical investigation?
Clinical investigation means any experiment in which a drug is administered or dispensed to, or used involving, one or more human subjects. For the purposes of this part, an experiment is any use of a drug except for the use of a marketed drug in the course of medical practice. (21 CFR 312.3(b)) Note: The fact that a clinical investigation can be any experiment in which a drug is administered or dispensed to, or used involving, one subject distinguishes it from the Common Rule’s definition of research under 45 CFR 46. 102, which requires “a systematic investigation, including research development, testing, and evaluation, designed to develop or contribute to generalizable knowledge.”
What is a sponsor?
Sponsor means a person who takes responsibility for and initiates a clinical investigation. The sponsor may be an individual or pharmaceutical company, governmental agency, academic institution, private organization, or other organization. The sponsor does not actually conduct the investigation unless the sponsor is a sponsor-investigator. (21 CFR 312.3(b))
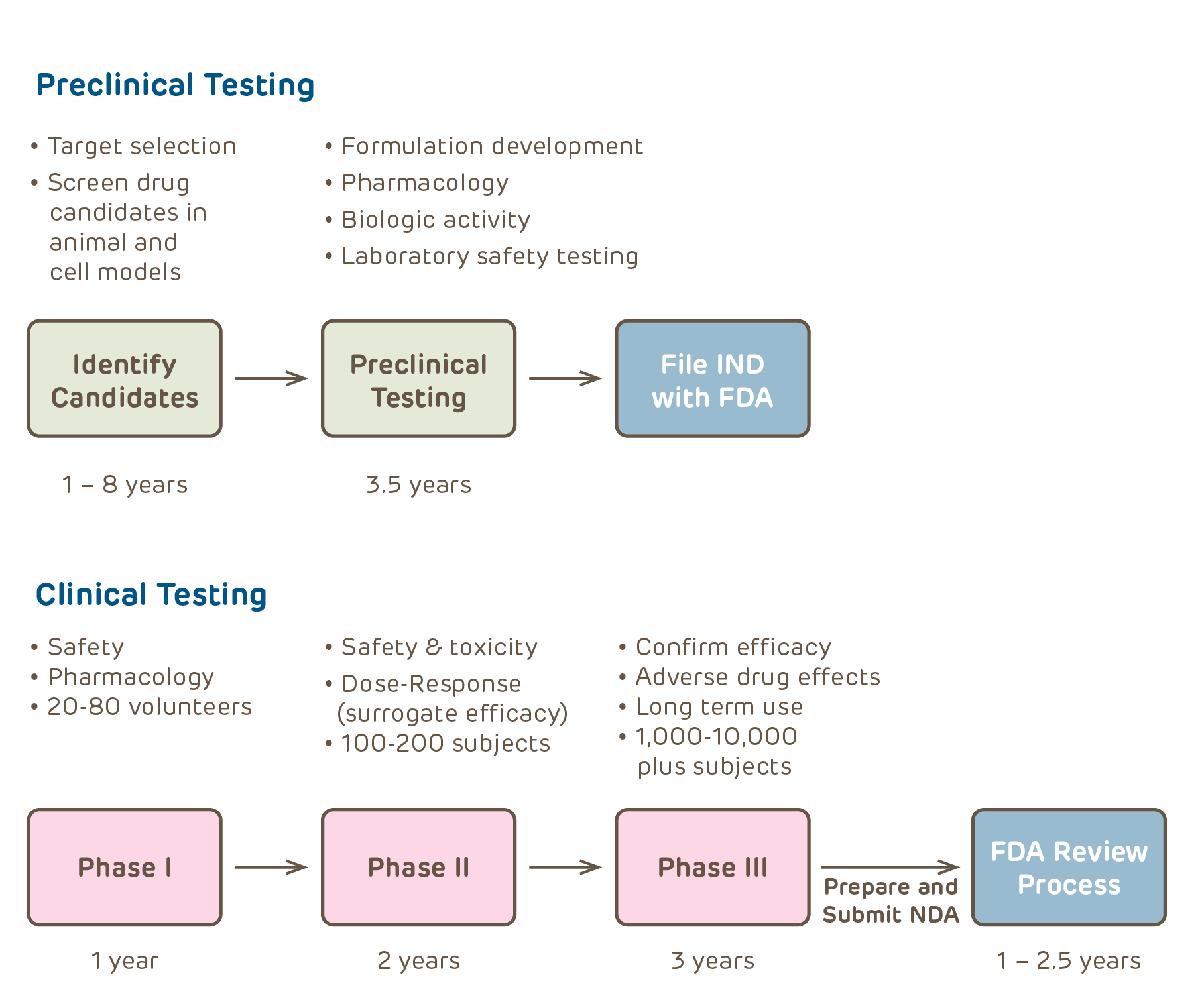
This section provides comprehensive information for investigators that helps determine whether or not an IND is needed for the proposed study, the process of applying for an IND with the FDA, and the responsibilities of investigators who hold INDs.
Frequently Asked Questions
-
I want to use a marketed drug off label in clinic. Do I need to submit to the IRB or the FDA?
Good medical practice and the best interests of the patient require that physicians use legally available drugs, biologics and devices according to their best knowledge and judgement. If physicians use a product for an indication not in the approved labeling, they have the responsibility to be well informed about the product, to base its use on firm scientific rationale and on sound medical evidence, and to maintain records of the product's use and effects. Use of a marketed product in this manner when the intent is the "practice of medicine" does not require the submission of an Investigational New Drug Application (IND), Investigational Device Exemption (IDE) or review by an Institutional Review Board (IRB). However, other institutional oversight requirements (e.g. Therapeutic Standards Committee approval) may apply. See FDA Guidance on "Off-Label" and Investigational Use Of Marketed Drugs, Biologics, and Medical Devices.
-
I want to conduct a clinical investigation using a marketed drug off label. Do I need to submit an IND application to the FDA?
Maybe.
Whether an IND is needed to conduct a clinical investigation of a marketed drug primarily depends on the intent of the investigation and the degree of risk associated with the use of the drug in the investigation. A clinical investigation of a marketed drug is exempt from the IND requirements if all of the criteria for an exemption in § 312.2(b) are met:- The drug product is lawfully marketed in the United States.
- The investigation is not intended to be reported to FDA as a well-controlled study in support of a new indication and there is no intent to use it to support any other significant change in the labeling of the drug.
- In the case of a prescription drug, the investigation is not intended to support a significant change in the advertising for the drug.
- The investigation does not involve a route of administration, dose, patient population, or other factor that significantly increases the risk (or decreases the acceptability of the risk) associated with the use of the drug product (21 CFR 312.2(b)(1)(iii)).
- The investigation is conducted in compliance with the requirements for review by an IRB (21 CFR part 56) and with the requirements for informed consent (21 CFR part 50).
- The investigation is conducted in compliance with the requirements of § 312.7 (i.e., the investigation is not intended to promote or commercialize the drug product).
See FDA Guidance on Determining Whether Human Research Studies Can Be Conducted Without an IND. and IND Exemptions for Studies of Lawfully Marketed Drug or Biological Products for the Treatment of Cancer
-
What are the IRB’s responsibilities in the selection of clinical investigators and research sites, and determining if an investigational new drug application (IND) or investigational device exemption (IDE) is required?
The regulations at 21 CFR 56.107(a) require that an IRB "...be able to ascertain the acceptability of the proposed research in terms of institutional commitments and regulations, applicable law, and standards of professional conduct and practice..." In addition, the regulations at 21 CFR 56.111 require that an IRB determine that the proposed research satisfies the criteria for approval, including that "...risks to subjects are minimized...[and] reasonable in relation to anticipated benefits, if any, to subjects...".
Qualification of investigators
The IRB needs information about the qualifications of the investigator(s) to conduct and supervise the proposed research. The IRB may also need to assess the investigator’s training and experience specifically related to the proposed study, particularly if the proposed research involves higher risks, vulnerable subjects, or novel technologies. In its assessment, the IRB may also rely on the clinical investigator’s medical department/division for information about the clinical investigator’s qualifications, or require a statement regarding the investigator’s qualifications from the chair of the investigator’s department/division.
Adequacy of the research site
The IRB needs to assess the site where the proposed research will take place to ensure it can adequately execute the protocol requirements. Depending upon the nature and risks of the proposed research and the IRB's prior knowledge of, or relationship to, the institution or other site at which the research will take place, this may be relatively simple and straightforward or it may entail a more involved assessment.
Verifying the determination of whether an IND or IDE is required for an FDA-regulated investigation
It is the sponsor’s responsibility to make an initial determination on whether a clinical investigation meets the criteria for an IND exemption. At CHOP, the IND-IDE Support Program can help a CHOP sponsor determine whether a specific study meets these criteria. The sponsor needs to include their determination (and the basis for that determination, including any supporting documentation) to the IRB with their protocol submission in eIRB. The IRB will, based on the scientific literature and generally known clinical experience (as presented by the investigator), either agree or disagree that there is no significant increase in the risk (or decrease in the acceptability of the risk) associated with the use of the drug product.
If the IRB agrees with the sponsor’s determination, no IND submission to the FDA is necessary.
If the IRB disagrees or has concerns that cannot be addressed by the sponsor, the sponsor will need to submit to the FDA. The FDA will then either issue an exempt determination or decide that an IND is required. The FDA’s decision is final.
See FDA Guidance on IRB Responsibilities for Reviewing the Qualifications of Investigators, Adequacy of Research Sites, and the Determination of Whether an IND/IDE is Needed. -
I want to study a dietary supplement in a clinical investigation. Do I need to submit an IND application to the FDA?
It depends.
Under the Dietary Supplement Health and Education Act of 1994 (DSHEA), a dietary supplement is defined, in part, as a product taken by mouth that is intended to supplement the diet and that contains one or more dietary ingredients.
Under DSHEA, a dietary supplement is not considered a drug if the intended use for which it is marketed is only to affect the structure or any function of the body (i.e., not intended to be used for a therapeutic purpose).
Similarly, whether an IND is needed for a clinical investigation evaluating a dietary supplement is determined by the intent of the clinical investigation. If the clinical investigation is intended only to evaluate the dietary supplement’s effect on the structure or function of the body, an IND is not required.
However, if the clinical investigation is intended to evaluate the dietary supplement’s ability to diagnose, cure, mitigate, treat, or prevent a disease, an IND is required under 21 CFR 312.
See FDA Guidance on Determining Whether Human Research Studies Can Be Conducted Without an IND. -
I am the sponsor. Can I charge for an investigational drug?
Maybe.
A sponsor can only recover the direct costs of making a drug available to subjects in a clinical trial — that is, those costs that are specifically and exclusively attributable to providing the drug to clinical trial subjects (21 CFR 312.8(d)(1)). These include costs to manufacture the drug in the quantity needed to conduct the clinical trial for which charging has been authorized or costs to acquire the drug from another source, including costs to ship and handle (e.g., store) the drug.
Under 21 CFR 312.8(d)(3), to support its calculation of recoverable costs, a sponsor must provide documentation to the FDA describing recovery of direct costs and, if applicable, describing certain additional costs that may be recovered for intermediate-size patient population expanded access uses or treatment INDs or protocols. This documentation must be accompanied by a statement that an independent, certified public accountant has reviewed and approved the calculations (21 CFR 312.8(d)(3)).
Once an authorization to charge has been obtained from the FDA, it needs to be attached to the eIRB application for the respective study. The consent form should reflect that the sponsor(-investigator) intends to charge for the drug.
See FDA Guidance on Charging for Investigational Drugs Under an IND — Questions and Answers. -
What is expanded access and how is it different from clinical trials?
Expanded access refers to the use of an investigational drug when the primary purpose is to diagnose, monitor, or treat a patient’s disease or condition.
The terms expanded access, access, and treatment use are used interchangeably. The terms compassionate use and preapproval access are also occasionally used in the context of the use of an investigational drug to treat a patient. Although these terms have been used informally in the United States and are used outside the United States, they are not defined or described in FDA regulations.
The main distinction between expanded access and the use of an investigational drug in the usual studies covered under an IND is that expanded access uses are not primarily intended to obtain information about the safety or effectiveness of a drug. Expanded access to an investigational drug can only be provided under a treatment IND or protocol if the sponsor is actively pursuing, with due diligence, marketing approval of the drug for the expanded access use.
Under FDA’s current regulations, there are three categories of expanded access:- Expanded access for individual patients, including for emergency use (21 CFR 312.310).
- Expanded access for intermediate-size patient populations (generally smaller than those typical of a treatment IND or treatment protocol — a treatment protocol is submitted as a protocol to an existing IND by the sponsor of the existing IND) (21 CFR 312.315).
- Expanded access for widespread treatment use through a treatment IND or treatment protocol (designed for use in larger patient populations) (21 CFR 312.320).
See FDA Guidance on Expanded Access to Investigational Drugs for Treatment Use — Questions and Answers.
-
I want to treat one of my patients with an investigational drug. I need to do this as soon as possible. Do I need to submit to the IRB or FDA?
Except for emergency expanded access use when there is not sufficient time to secure prospective IRB review, an investigator treating a patient with an investigational drug under expanded access is responsible for obtaining IRB review and approval consistent with 21 CFR part 56 before treatment with the investigational drug may begin.
Generally, the investigator needs to contact the IRB to find out whether the IRB has sufficient time to review. If there is insufficient time for the IRB to review, FDA authorization is still required (21 CFR 312.310(d)), but it is not necessary to wait for IRB approval to begin treatment. However, the IRB must be notified of the emergency expanded access use within 5 working days of emergency use (21 CFR 56.104(c)).
For further guidance, go to Single Patient Expanded Access or Emergency Use and contact CHOP’s IND/IDE Support Program.
Also see the FDA Guidance on Expanded Access to Investigational Drugs for Treatment Use — Questions and Answers. -
Do I need to obtain consent from a subject who receives an investigational drug through expanded access?
Yes.
Expanded access to an investigational drug for treatment use, including emergency use, requires informed consent as described in 21 CFR part 50. Investigators treating a patient(s) with an investigational drug under expanded access are responsible for ensuring that the informed consent requirements of part 50 are met (21 CFR 312.305(c)(4)). One of the purposes of informed consent is to ensure that the patient is informed that he/she will be treated with an investigational product and that there may be uncertainty about the safety and effectiveness of the product.
See FDA Guidance on Expanded Access to Investigational Drugs for Treatment Use — Questions and Answers. -
I am using a placebo in a clinical investigation. Does the placebo need an IND?
Usually not.
If the placebo consists of inactive ingredients (e.g. excipients considered to be generally recognized as safe (GRAS)), an IND is not required. A clinical investigation involving use of a placebo is exempt from the requirements of this part if the investigation does not otherwise require submission of an IND (21 CFR 312.2(b)(5)).
See FDA Guidance on Exploratory IND Studies.