HOW CAN WE HELP YOU? Call 1-800-TRY-CHOP

In This Section
Search IRB Resources
Informed Consent and Documentation
Published on Jun 14, 2022 · Last Updated 1 month 1 week ago
The Informed Consent Process
The Nuremberg code lists this requirement as its first ethical principle. Obtaining the informed consent of the subject is therefore fundamental to ethical conduct of human subjects research. The informed consent of the subject demonstrates respect for persons.
Respecting research participants shows respect for their autonomy and right to self-determination and treats requires the investigator to treat like individuals alike. To obtain the informed consent of a participant the following conditions must be met:
- the subjects must be informed, it is insufficient to simply present the information to the subject if they do not comprehend that information.
- they must have the capacity - legally, developmentally, intellectually - to understand the purpose of the research, the nature of the procedures, the potential risks and benefits inherent in the choices presented and the voluntary nature of their participation;
- they must be competent to freely exercise their choices.
Informed consent is not a document, it is a process that begins with recruitment and continues until the subject's participation in the research is completed. Even then, if the subject's private identifiable information continues to be used, the investigator has an obligation to inform the subject of changes in the research.
The consent process involves providing the information that a reasonable person would want to know in language that is understandable to the subject. Understandable means at a grade level that they can understand and in their native language. If the prospective subject can not demonstrate that they comprehend the basic elements of consent then the investigator should not enroll the subject.
Consent is usually documented on a written consent form but the process could take place via some other media (e.g. video, DVD, computer, etc.). Under some circumstances the IRB can waive the requirement for documentation (see Waiver of Documentation of Consent) of consent.
Consent Decision Tree
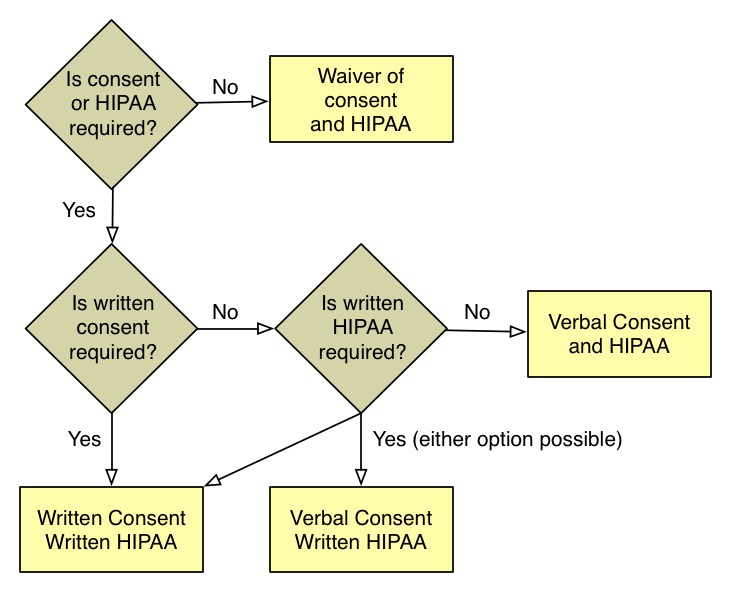
The IRB must determine whether or not consent and HIPAA authorization are required and if they are, whether or not they need to be documented. The figure above shows the decision process for consent and HIPAA and includes decisions about the following:
- whether or not informed consent is required;
- if the study qualifies for a waiver of consent, whether or not it qualifies for a waiver or HIPAA authorization;
- if it is required, whether the study qualifies for a waiver of documentation of consent (e.g. verbal consent) or requires a written consent form;
- if the research qualifies for a waiver of documentation or an alteration of consent, whether or not it also qualifies for an alteration of HIPAA (e.g. verbal HIPAA authorization); and
- if children are subjects, whether or not assent is required and if it is, how it will be documented.
Parental Permission and Assent
Children have neither the legal status nor the development capacity to understand many of the issues inherent in providing informed consent. Since no one can consent for another, a substituted process of alternative protections are in place for children. Parents must provide their permission for a child's participation in research just as they provide permission for clinical care and the child must assent.
The regulatory requirements for parental permission are on the Children in Research page. The requirements for assent of children are outlined on the Assent of Children page. Waiver of the requirement for assent and Waiver of the requirement for parental permission are both possible under some circumstances.
During adolescence minors develop an increasing capacity to make informed choices. By age 14 years, teens understand as much (or as little) information about proposed clinical research studies as adults do. Under some circumstances minors are not considered to be children under the research regulations. Whenever they are permitted under state law to consent to their own treatment, they may consent to their own research participation.
Sophie's Science Project. What is Medical Research?
Sophie's Science Project is a comic-book style to help parents and children learn about clinical research. It was developed by Athos Bousvaros MD, MPH and Susan Kornetsky, MPH at Boston Children's Hospital who created it to educate parents and children about medical research and the informed consent process. The project also had support from Cincinnati Children's Hospital and Medical Center and The Children's Hospital of Philadelphia.
Depending on the resolution and size of the device being used, it may be necessary to view Sophie's Science Project in full screen mode to read it. Paper copies of Sophie's Science Project are available by calling the IRB Office or the CRSO.
Subjects Who Cannot Consent for Themselves
Clinicians and investigators must establish procedures to determine who is legally permitted to grant permission (consent) from children and adults with diminished capacity who are unable to consent for themselves. investigators must conform to CHOP Policy on Consent. This policy applies not only to consent for treatment but also to consent for research participation.
The following definitions apply:
Parent:
Means biologic or adoptive.
Guardian:
Means an individual authorized under state or local law to consent on behalf of the child.
Legally Authorized Representative:
Means the individual designated in accordance with state law to consent for an adult who is incapable of providing consent.
Wards of the State/ Foster Children:
Foster parents may NOT consent for treatment or research participation for minor children. Only when a legally authorized individual grants permission, may a foster child participate in research.
See IRB SOP 504: Research Involving Children and the webpages on Children in Research for more information.
Obligations for Obtaining Permission/Informed Consent for Child Participants
In conformance with the additional protections for children (Subpart D), the IRB assigns a risk-benefit category to all research involving children as subjects. The risk category is listed in the approval letter and determines whether one or both parents/guardians must grant permission to allow their child to participate in research.
Children in Research provides additional details regarding the four risk-benefit categories.
Participants, Parents/Guardians Who Are Non-English Speaking
If the subject or their parents/guardians are non-English speaking, either the consent form must be translated into the native language or a Short Form consent process must be used. See the IRB webpage on Short Form Consent for more details.
Witnesses to the Short Form Consent Process
A witness (fluent in both languages) to the consent process is only required, when the subject's parent/guardian are illiterate or if a Short Form Consent process is used.
Informed Consent Requirements for Screening Procedures
The IRB may approve certain screening procedures without requiring prior informed consent (or parental permission and assent) of the prospective subject or the subject’s legally authorized representative under certain conditions. For additional information, see Recruitment vs Screening.
Assent of Children
Learn more about the requirements and assent of children.