HOW CAN WE HELP YOU? Call 1-800-TRY-CHOP

In This Section
Search IRB Resources
Incidental Findings
Published on Jun 14, 2022 · Last Updated 1 year 2 months ago
"An IF is a finding concerning an individual research participant that has potential health or reproductive importance and is discovered in the course of conducting research but is beyond the aims of the research. This means that IFs may be on variables not directly under study and may not be anticipated in the research protocol."
Wolf et al. J Law Med & Ethics 32:219, 2008
Incidental findings in research are becoming increasingly common due to the advances in imaging and genetics research. Investigators must consider the possibility of IFs as part of their assessment of the risks and benefits of research participation and must have a plan for reporting results.
Examples:
- A possible brain tumor or a vascular malformation found on a functional MRI scan;
- A laboratory test abnormality found as part of screening for a clinical trial or for baseline physiologic data on a "healthy" control subject;
- A possible genetic abnormality or risk factor;
- Discovery of non-paternity determined by genetic testing of parents (up to 10% in some studies)
- Discovery that a subject may be suicidal from the results of a quality of life survey (for more on suicidality see the Behavioral Research page.
Which results are clinically significant?
There is considerable debate in the research ethics community about which results are clinically significant. At a minimum any results communicated to subjects should meet the following criteria:
-
Analytic validity:
this means that the test was performed in a laboratory with established procedures to ensure reproducibility. In keeping with CHOP clinical guidelines, IRB SOP 903: Reporting Test Results and Incidental Findings interprets this to mean performed in a CLIA-certified lab. However, the FDA reported on July 31, 2014 that they were issuing "a final guidance on the development, review and approval or clearance of companion diagnostics, which are tests used to identify patients who will benefit from or be harmed by treatment with a certain drug" and they were intending "to publish a proposed risk-based oversight framework for laboratory developed tests (LDTs), which are designed, manufactured and used within a single laboratory going to end its enforcement discretion related to lab-developed tests." This latter action could especially impact the use of genetic tests. -
Known clinical significance and utility:
which means that there are important implications to the participant's health and well-being which justify the risks of anxiety and concern that will be raised by knowing the results. This requires that there are effective preventive measures, treatments or interventions currently available.
What are the investigator's obligations?
Investigators are obligated to include the following in their research plans (usually the protocol):
- A plan for identifying and assessing which IF are of likely clinical significance. Since IF may be outside the investigator's expertise, this could include a plan for obtain clinical expertise from non-investigators.
- Identify which results will be reported to subjects and the circumstances of the communications (who, when, where and what). This involves deciding which results are of known clinical significance and utility and this may change over the course of the research.
- Develop a plan for further care. This could include provision of care by the investigator, referral to another clinic, physician or provider or information about alternative resources for obtaining care.
The following information should be in the consent document:
- The plan for reporting IF information;
- Inclusion of "any foreseeable risks" and "any benefits" that derive from reporting IF as required by regulation. IF that have uncertain clinical implications and where there are no known treatments or interventions may cause subjects undue concern, anxiety and worry. IF that identify a major health problem, could be of tremendous benefit.
- Inclusion of an opt-out from knowing the results of research tests.
- It may not be possible (due to e.g. time constraints) to discuss all the implications of returning results at the time of initial consent. In this case, investigators should strongly consider discussing the options for a return of results separately with the participant (e.g. with a genetic counselor). The IRB has developed a consent addendum to facilitate such a discussion and allow participants to choose which results to have returned to them (Return of Results Consent Addendum).
All clinically significant research results must be recorded in the CHOP Medical Record.
- Refer to the CHOP Research Institute Policies to determine which results must go in the medical record.
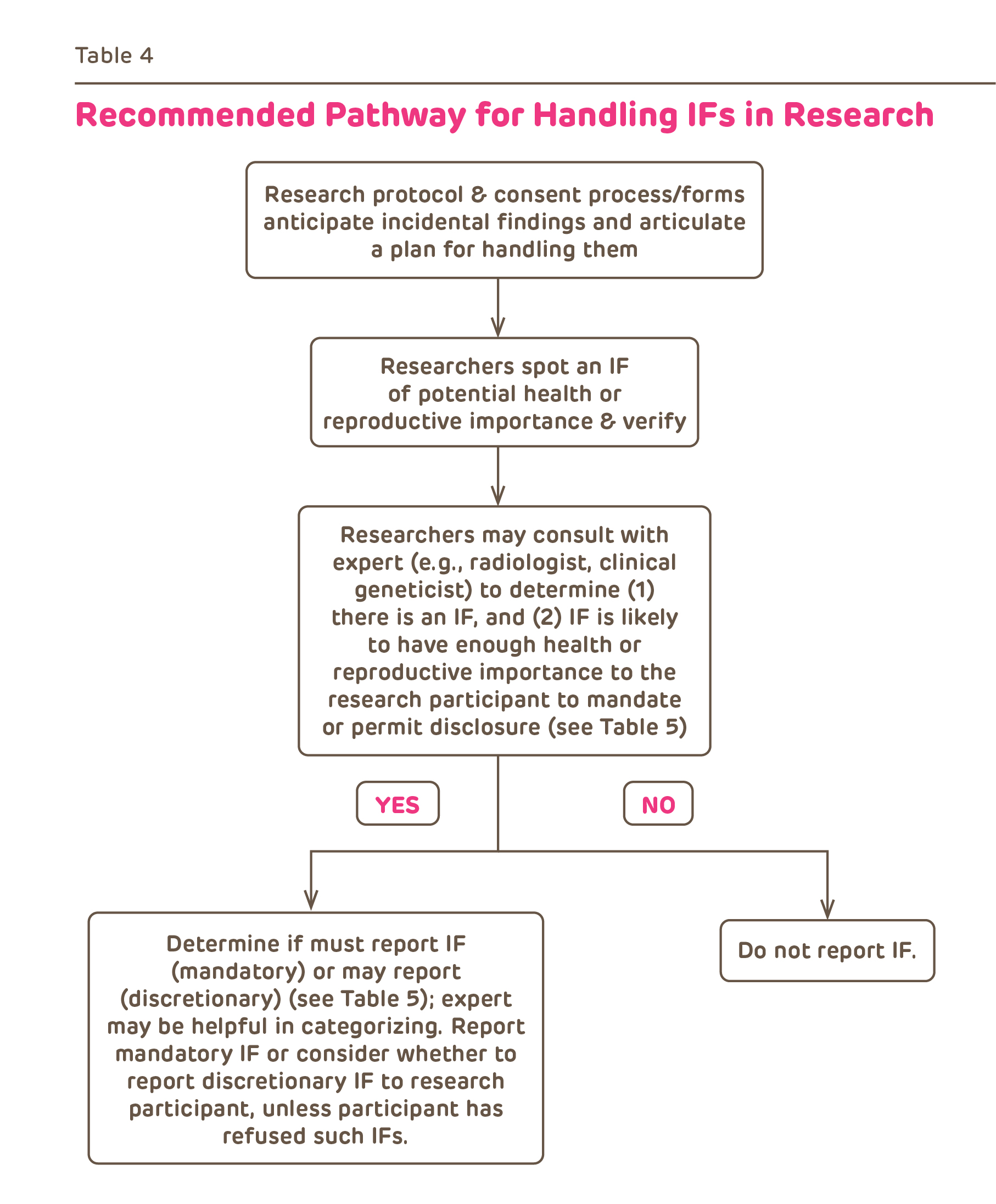
From Wolf et al. J Law Med & Ethics 32:234, 2008
Recommendations for Returning Research Results
| Comparison of Recommendations on Returning Individual Research Results | |
|---|---|
|
National Bioethics Advisory Commission (NBAC) |
Return results only if:
|
|
Centers for Disease Control |
Criteria for returning individual results in population-based genetic research: "When the risks identified in the study are both valid and associated with a proven intervention for risk reduction, disclosure may be appropriate." |
|
National Heart, Lung, and Blood Institute (NHLBI, 2010) |
Criteria for returning individual genetic results (abbreviated):
|
|
National Research Council & Institute of Medicine (NRC & IOM) |
In human embryonic stem cell research, the duty to report individual research results "depends in large part on the reliability of the findings and the significance of the information to human health." "CLIA regulations do not permit the return of research results to patients or subjects if the test were not conducted in a CLIA-approved laboratory." |
|
National Human Genome Research Institute (NHGRI) |
Upon their request, "[r]esearch participants should have access to experimental research data except when...[t]he reserach results are of unproven clinical validity, and the IRB has judged that there is no benefit to the research subjects." |
| From Wolf et al. J Law Med & Ethics 32:219, 2008 | |
References
- Wolf SM, et. al. Managing Incidental Findings in Human Subjects Research
- Wolf SM, et al. The Law of Incidental Findings
- Dickert N, Wendler D. Ancillary Care Obligations of Medical Researchers
- Jarvik GP. Return of Genomic Results to Research Participants: The Floor, the Ceiling, and the Choices In Between
- Fabsitz RR, et al: Ethical and Practical Guidelines for Reporting Genetic Research Results to Study Participants. Updated Guidelines From a National Heart, Lung, and Blood Institute Working Group. Circulation: Genomic and Precision Medicine. 2010;3:574-580
- Abdul-Karim R, et al: Disclosure of Incidental Findings From Next-Generation Sequencing in Pediatric Genomic Research. Pediatrics 2013;131:564–571