HOW CAN WE HELP YOU? Call 1-800-TRY-CHOP

In This Section
Search IRB Resources
Do I need a protocol and if so, how do I write a protocol?
Published on Jun 14, 2022 · Last Updated 1 year 6 months ago
Start Out on the Right Foot
If you don't start looking for a biostatistician co-principal investigator the same day that you start formulating your study question, you are a fool, and deserve neither funding nor a valid answer.
- David Sackett
Clinical Epidemiology: How to Do Clinical Practice Research (Table 4.2)
If authors do not consider key methodological features at the design and execution phases of their study, they will have difficulties in improving the paper at the later scientific phases.
- Erik Cobo, et al.
Federal regulations and international standards require that IRBs review and approve human subjects research prior to initiation of a clinical research study. To do that, the IRB must make a number of determinations in order to ensure that the study meets all of the regulatory requirements for approval. The protocol serves many purposes, one of which is to provide the IRB with all of the necessary information to ensure that the study will follow principles of sound scientific design and that the research will be conducted in accordance with federal regulations, state law and CHOP IRB and CHOP Research Institute policies.
Many IRB submissions cut and paste information from the investigator's grant. This information provides much more scientific background information than is appropriate for a protocol and does not provide sufficient information regarding conduct and execution of the study. A protocol is a recipe that should enable anyone knowledgeable in clinical study execution to conduct the study; it is specific and detailed to ensure that the study will have fruitful results. At the other extreme, many protocols contain virtually no information and may be less than a single page. Many of the important scientific and ethical issues are not dealt with at all.
The Office of Human Research Protections (OHRP) has frequently cited IRBs for failure to require complete protocol information. For example, OHRP has cited IRBs for reviewing only minimal information regarding (a) risks to subjects and how they are minimized; (b) subject recruitment and enrollment procedures; (c) the equitable selection of subjects; (d) provisions to protect the privacy of subjects and maintain the confidentiality of data; and (e) additional safeguards to protect the rights and welfare of subjects who are likely to be vulnerable. See OHRP Compliance Findings for their most frequently cited compliance issues.
Do I need a protocol?
There are several types of IRB submissions for which the IRB does not require a protocol; the investigator can complete a textbox or checkboxes to describe their project. Examples include:
- Studies which qualify for exemption;
- Requests for determination of the need for IRB review;
- Submissions of HIPAA Attestations
Resources to Assist with Protocols
A Well-Written Protocol: Key to Rapid IRB Approval
Facilitating IRB Approval
Advice for writing protocols isn't strictly speaking, the IRB's responsibility. However, in reviewing the reasons for deferral or disapproval of research applications, the CHOP IRB has identified poorly written protocols as a major factor. The IRB has found that providing protocol templates and resources related to protocol writing reduces both the investigator's and the IRB's work and enables investigators to gain IRB approval more quickly.
The protocol serves as the recipe for the conduct of the research activity. It needs to communicate all of the information that the following groups need to conduct their part of the study: the investigator and the investigative team, the data manager, the statistician and the IRB or other review body. Therefore, the protocol must include the scientific rationale to justify the conduct of the study, the information necessary to conduct the study, the plan for managing and analyzing the data, and a discussion of the research ethical issues relevant to the research. A complete, well-written protocol should address all scientific and research ethics issues so that the IRB does not need to request clarifications or to seek additional information.
Key Features of a Protocol
- Sufficient scientific rationale to justify conduct of the study
- Details sufficient for subsequent repetition by other investigators, publication in a quality scientific journal and ultimately, inclusion in a meta-analysis;
- Scientifically sound and appropriate study design
- Study population clearly defined (inclusion and exclusion criteria)
- All procedures sufficiently specified such that:
- Study coordinator can ensure study visits completed correctly;
- Measurements specified in sufficient detail to ensure that data manager could create case report forms; and
- Study team can perform measurements in a consistent, reproducible manner.
- Analysis plan and sample size complete, including methods for:
- Allocation assignment and blinding methods;
- Blinding of treatments;
- Analysis of primary and secondary endpoints in sufficient details for scientific review and to allow a statistician, otherwise unfamiliar with the study to repeat the analysis;
- Sufficient detail for journal publication (compliant with consensus reporting guidelines)
- Cogent and complete discussion of ethical issues that is sufficient to address all of the regulatory requirements for approval
Good Clinical Practice Protocol Standards
The appropriate study design should be chosen to provide the desired information...
Appropriate comparators should be utilized and adequate numbers of subjects included to achieve the study objectives. Primary and secondary endpoints and plans for their analyses should be clearly stated.
ICH General Considerations for Clinical Trials: E8
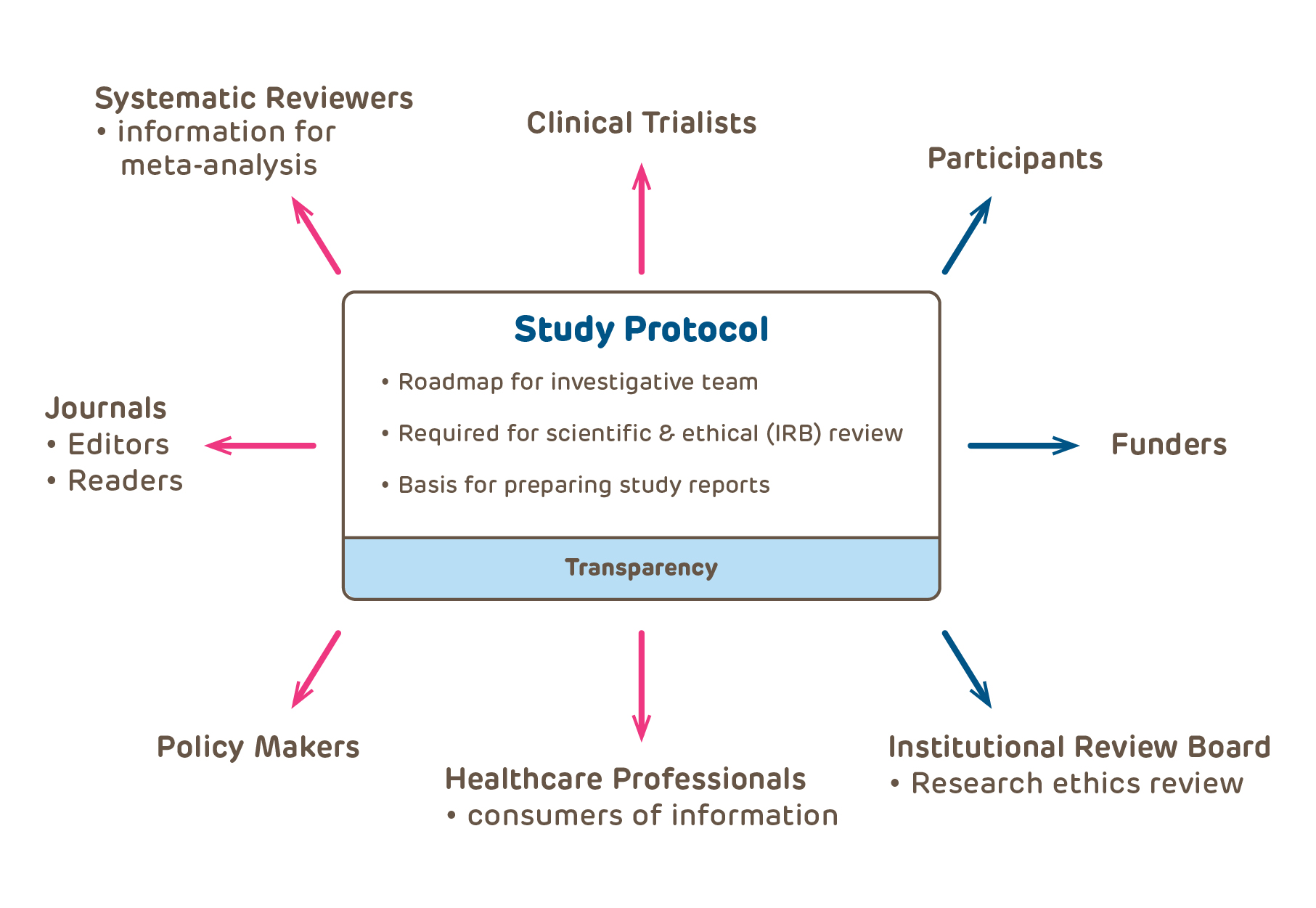
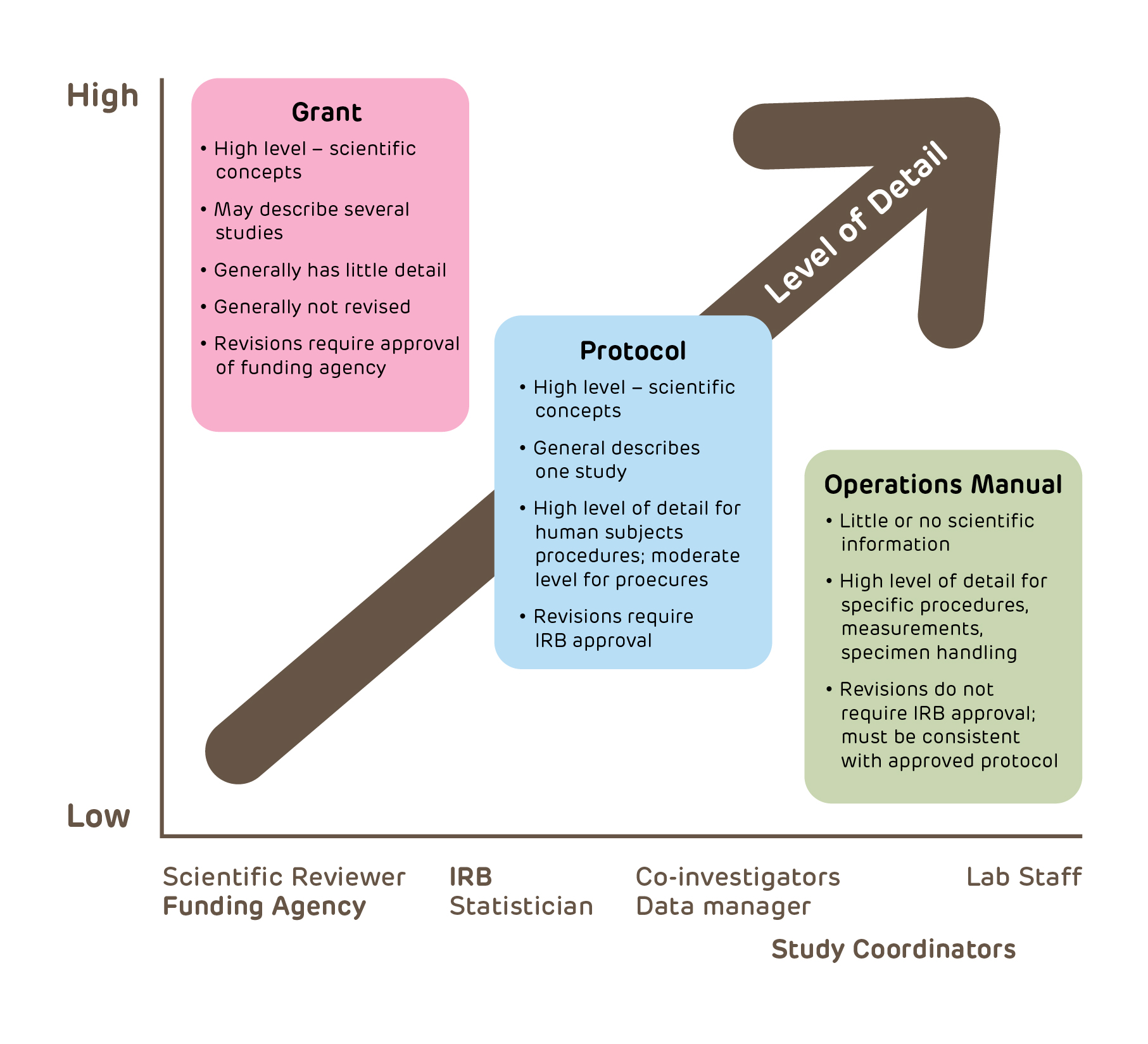
How is a protocol different than a grant?
Many IRB submissions cut and paste information from the investigator's grant. This information provides much more scientific background information than is appropriate for a protocol and does not provide sufficient information regarding conduct and execution of the study. A protocol is a recipe that should enable anyone knowledgeable in clinical study execution to conduct the study; it is specific and detailed to ensure that the study will have fruitful results. At the other extreme, many protocols contain virtually no information and may be less than a single page. Many of the important scientific and ethical issues are not dealt with at all.
Research Grant versus Study Protocol:
Grant (and if applicable the Investigators Brochure): contains all scientific basis for proposed research including background, hypothesis and general description of methods;
versus
Protocol: step-by-step details of study objectives, design, procedures, measurements, management and analysis, as well as a discussion of relevant research ethics issues
Textbook on kitchen chemistry which explains why bread rises;
versus
Recipe containing the instructions for baking bread that includes a list of ingredients, measurements, and step-by-step procedures to be followed.
Definitions: Screened, Enrolled, and Evaluable
To ensure that enough subjects participate to achieve the study objectives, the protocol should estimate the number of evaluable subjects needed. Some subjects will be recruited by decline to participate, some will consent but will be screen failures or will drop out. Only subjects who meet certain criteria (see below) will ultimately be included in the analysis of the study. It is important to estimate how many subjects will be lost during screening and during the study in order to have enough subjects at the end.
Screened means assessed for eligibility for the study. If the investigator must interact with the subject to obtain information to compare against the inclusion/exclusion criteria, informed consent may need to be obtained before screening commences:
- The IRB may approve certain screening procedures without requiring prior informed consent (or parental permission and assent) of the prospective subject or the subject’s legally authorized representative under certain conditions. For additional information, see Recruitment vs Screening. Note that, even if consent does not need to be obtained for screening procedures, HIPAA Authorization may still need to be obtained if the requested data/stored biospecimens include PHI (the IRB's responsibilities related to HIPAA are described in more detail in the IRB's Role in HIPAA).
- If screening procedures are not limited to the procedures outlined on Recruitment vs Screening, then informed consent must be obtained before screening commences.
- If the investigator simply informs the prospective subject about the trial, this is recruitment not screening.
Enrolled means all subjects who have consented to screening (if applicable) or participation in the main study. If the subject is a screen failure (ineligible), they are still enrolled. Subjects who fail to get the study intervention or to complete the required study procedures are still enrolled.
Evaluable means all subjects whose data can be used in the final analysis data set. Subjects may be evaluable for some endpoints but not others. For example, if 4 blood samples are needed per subject to be considered evaluable in a PK study, all those who get the study drug but provide fewer specimens would be evaluable for safety outcomes but not the primary outcome (PK).
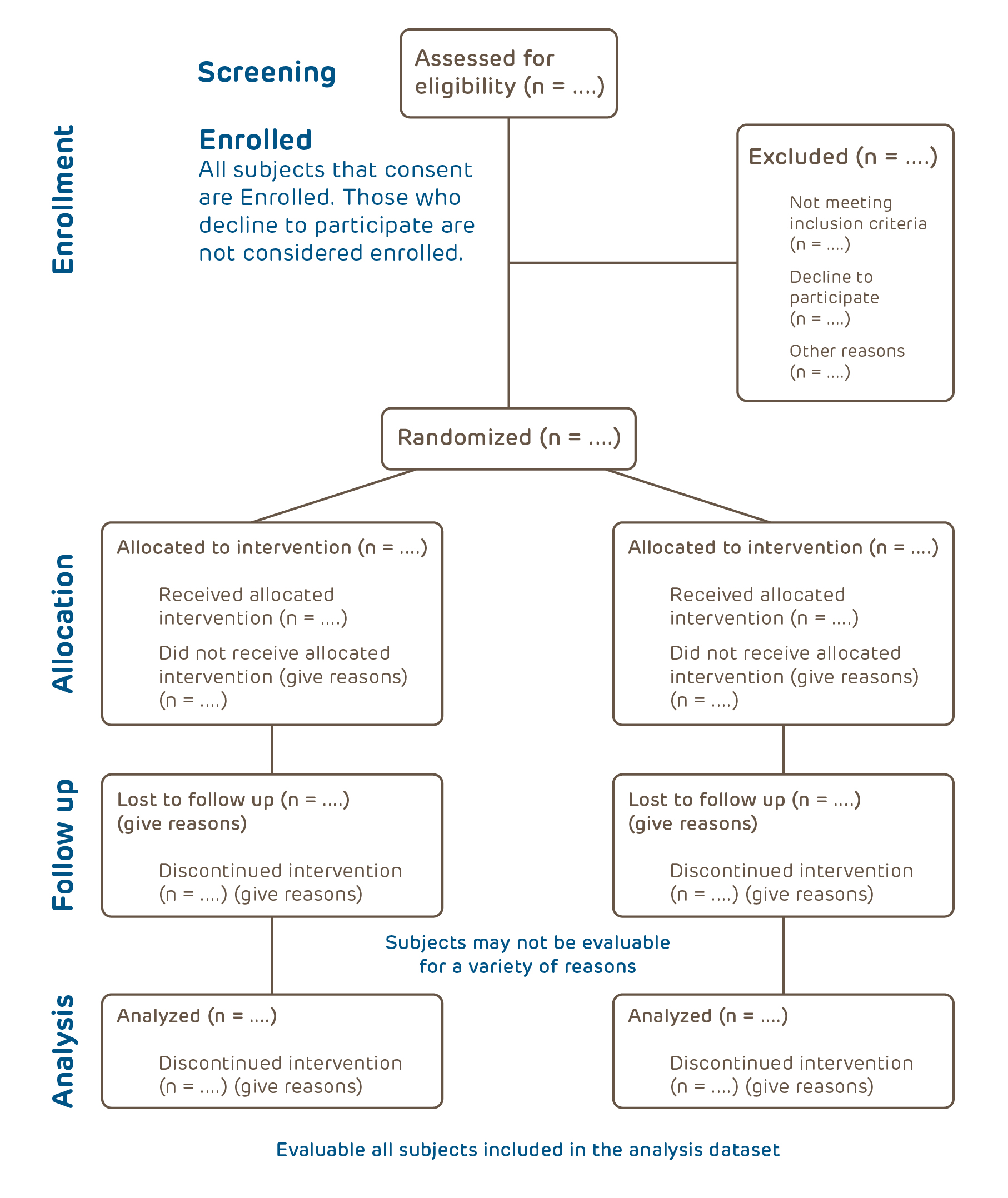
The figure above is the required study flow diagram from the CONSORT guidelines. All subjects should be accounted for in the final study report and publication.
Frequently Asked Questions
-
I'm an investigator for a multi-center study; do I need to write a CHOP-specific protocol?
The IRB doesn't want and will not accept a CHOP-specific protocol for multi-center studies (unless you are the PI and this will be the protocol used at all sites). The IRB will accept one and only one protocol to serve as the official protocol for multi-center research. The IRB is obligated to ensure that the research uses sound scientific design. When there is more than one protocol the IRB cannot be certain if all centers will include all of the required procedures or carry them out the same way. If there is a need to describe study activities at CHOP than aren't well-described in either the protocol or the in the eIRB application, a protocol addendum may be created. Provided that the protocol is well-written, this should rarely be necessary.
-
What is the difference between the number of subjects enrolled and the number evaluable?
The Enrolled total is the number of subjects who consent to participate in the study. For a retrospective study, it would be the number of charts reviewed or specimens received. The Evaluable number is the total of the subjects that reach the required endpoint for the study.
Example: 100 subjects might enroll, 10 fail screening, and 20 drop out before the final study visit that is required for assessing the primary efficacy endpoint. The enrolled number is 100, the evaluable number is 80 and 90 subjects will be included in the safety analysis.
Example: 24 subjects are enrolled in a PK study but only 18 provide enough blood samples for their data to used in estimating PK parameters. The evaluable number is 18 and the enrolled is 24. All 24 subjects will be part of the safety analysis.
-
I've enrolled 25 subjects but only 5 have completed the study; how many do I list on the continuing review form as evaluable?
During a study with multiple visits, the investigator will not know how many of the enrolled subjects will ultimately be evaluable. The evaluable total at the time of continuing review should be based on those who have completed the study or reached the study visit where the primary endpoint assessment is made.
-
I know how many evaluable subjects are needed from the power analysis; how do I estimate the enrolled number?
The investigator should estimate the number of subjects who will fail screening, drop-out or be unable to complete required study procedures. This number when added to the required evaluable number will give an estimate of the number who will need to be enrolled. If fewer subjects drop-out then anticipated, the study might wind up with more evaluable subjects then anticipated. However, under-estimation of the number of subjects who will need to enroll in order to achieve the required evaluable total is often the bigger problem. Under-estimation can result in protocol deviations and burdens the investigator and IRB with protocol amendments to increase the approved enrollment number.
-
How many subjects should I estimate for the CHOP target for multicenter studies?
The total evaluable number for all sites is determined by the study protocol. The IRB prefers that the investigator use the highest total that could conceivably be enrolled at CHOP. This avoids the need for protocol amendments just to increase the permissible enrolled number.