HOW CAN WE HELP YOU? Call 1-800-TRY-CHOP

In This Section
Devices
Published on Jun 14, 2022 · Last Updated 1 year 2 months ago
What is a medical device?
21 USC 321(h) The term ‘‘device’’ .... means an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including any component, part, or accessory, which is
- recognized in the official National Formulary, or the United States Pharmacopeia, or any supplement to them,
- intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals, or
- intended to affect the structure or any function of the body of man or other animals, and which does not achieve its primary intended purposes through chemical action within or on the body of man or other animals and which is not dependent upon being metabolized for the achievement of its primary intended purposes. The term ‘‘device’’ does not include software functions excluded pursuant to section 360j(o) of this title.
Types of Device Studies
What is an investigational medical device?
(g) Investigational device means a device, including a transitional device, that is the object of an investigation.
(h) Investigation means a clinical investigation or research involving one or more subjects to determine the safety or effectiveness of a device.
(i) Investigator means an individual who actually conducts a clinical investigation, i.e., under whose immediate direction the test article is administered or dispensed to, or used involving, a subject, or, in the event of an investigation conducted by a team of individuals, is the responsible leader of that team.
(p) Subject means a human who participates in an investigation, either as an individual on whom or on whose specimen an investigational device is used or as a control. A subject may be in normal health or may have a med- ical condition or disease.
By applying the definitions (left), it becomes clear that simply using a device as part of a study does not make the device investigational. For example, if an non-invasive, unapproved device is used to make measurements as part of a study, but the study is not about the device, then the device is not an investigational device for the purposes of that study. For example, if an investigator wishes to use an unapproved device to measure cerebral blood flow but the study is not about the device (not assessing the safety or effectiveness of the device), then for that study, the unapproved device would not be investigational. Whereas the device would be an investigational device when used in a study designed to assess the feasibility and safety of making cerebral blood flow measurements during cardiac surgery.
Following the regulatory pathway for devices logic below and definitions on the Device Regulation and Classification accordion below, the sponsor/investigator should first determine if the study meets the definition of a clinical investigation of an investigational device. If the study is a clinical investigation of an investigational device, then the investigator must determine how to apply the FDA's device regulations. The Investigational Device Exemptions (IDE) regulations (21 CFR 812) describe three types of device studies: (1) exempt investigations, (2) investigations of significant risk (SR) devices, and (3) investigations of nonsignificant risk (NSR) devices.
The regulatory requirements vary, depending on which of the three types of device studies are involved. Humanitarian Use Devices, when used in accordance with their label - under a Humanitarian Device Exemption (HDE) - are a special case and follow different regulations. These are discussed below.
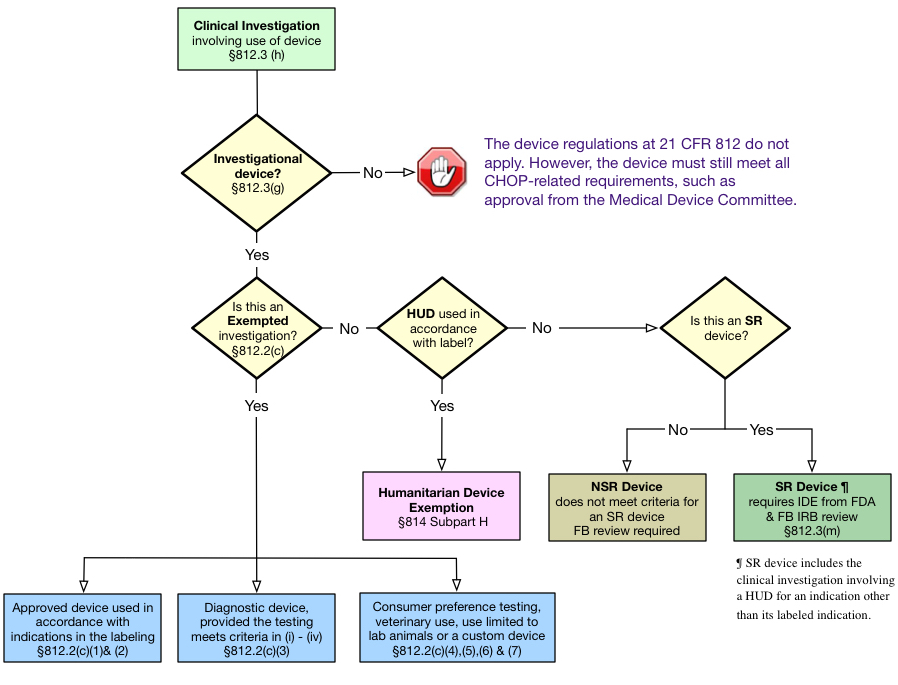
Exempted Investigations
With the exception of 21 CFR 812.119, the rest of the IDE regulations do not apply to device studies that meet the criteria for an exempted investigation under 21 CFR 812.2 (c). They also do not require the FDA, the sponsor, the investigator or the IRB to make an NSR or SR determination.
Exempted investigations include investigations involving one of the following:
- A device, other than a transitional device, in commercial distribution immediately before May 28, 1976, when used or investigated in accordance with the indications in labeling in effect at that time.
- A device, other than a transitional device, introduced into commercial distribution on or after May 28, 1976, that FDA has determined to be substantially equivalent to a device in commercial distribution immediately before May 28, 1976, and that is used or investigated in accordance with the indications in the labeling FDA reviewed under subpart E of part 807 in determining substantial equivalence.
- A diagnostic device, if the sponsor complies with applicable requirements in 809.10(c) and if the testing: (Comment: this is the most frequent type of exempted investigation submitted to the CHOP IRB.)
- Is noninvasive,
- Does not require an invasive sampling procedure that presents significant risk,
- Does not by design or intention introduce energy into a subject, and
- Is not used as a diagnostic procedure without confirmation of the diagnosis by another, medically established diagnostic product or procedure.
- A device undergoing consumer preference testing, testing of a modification, or testing of a combination of devices if the device(s) are legally marketed device(s) (that is, the devices have an approved PMA, cleared Premarket Notification 510(k), or are exempt from 510(k)) AND if the testing is not for the purpose of determining safety or effectiveness and does not put subjects at risk.
- A device intended solely for veterinary use.
- A device shipped solely for research on or with laboratory animals and labeled in accordance with 812.5(c).
- A custom device as defined in 812.3(b), unless the device is being used to determine safety or effectiveness for commercial distribution.
Significant Risk Devices
Under 21 CFR 812.3(m), an SR device means an investigational device that:
- Is intended as an implant and presents a potential for serious risk to the health, safety, or welfare of a subject;
- Is purported or represented to be for use supporting or sustaining human life and presents a potential for serious risk to the health, safety, or welfare of a subject;
- Is for a use of substantial importance in diagnosing, curing, mitigating, or treating disease, or otherwise preventing impairment of human health and presents a potential for serious risk to the health, safety, or welfare of a subject; or
- Otherwise presents a potential for serious risk to the health, safety, or welfare of a subject.
A SR device must follow all of the IDE Regulations at 21 CFR 812 and the study must be conducted under an Investigational Device Exemption (IDE) application approved by the FDA before the investigation begins.
Examples of SR Devices
- Catheters for General Hospital Use - except for conventional long-term percutaneous, implanted, subcutaneous and intravascular
- Surgical Lasers for use in various medical specialties
- Respiratory Ventilators and new modes of ventilation
- Cardiac Assist, Bypass and ECMO Devices
- Cardiac Pacemaker/Pulse Generators
- Cardiopulmonary Resuscitation (CPR) Devices
- Intravascular Stents, Percutaneous Transluminal Angioplasty Catheters, Heart Valves
- Implantable Prostheses and Vascular Devices (new devices)
- Dialysis Delivery Systems
- Sutures
- Infusion Pumps (implantable and closed-loop - depending on the infused drug)
Non-Significant Risk Devices
A NSR device is any device that doesn't meet the definition of a SR device. Studies involving an NSR need only follow the abbreviated device regulations at 21 CFR 812.2(b). It the sponsor's responsibility to provide the IRB with the following:
- their determination that the device is NSR,
- the reasons why it has come to this conclusion,
- information that needed to allow the IRB to evaluate the risk of using the device in the proposed study,
- a description of the device,
- the protocol and any other information that the IRB requires
At the time of its review the IRB must review the investigator/sponsor's NSR determination and either agree or disagree. In addition, the IRB must review and approve the study using the FDA's Human Subjects Research Regulations and IRB regulations at 21 CFR 50 and 56.
Examples of NSR Devices
- Contact Lens Solutions
- Conventional Gastroenterology and Urology Endoscopes and/or Accessories
- Conventional General Hospital Catheters
- Conventional Implantable Vascular Access Devices (Ports)
- Conventional Laparoscopes, Culdoscopes, and Hysteroscopes
- Electroencephalography (e.g., new recording and analysis methods, enhanced diagnostic capabilities, measuring depth of anesthesia if anesthetic administration is not based on device output)
- Externally Worn Monitors for Insulin Reactions
- Functional Non-Invasive Electrical Neuromuscular Stimulators
- General Urological Catheters (e.g., Foley and diagnostic catheters) for short term use (< 28 days)
- Low Power Lasers for treatment of pain
- Magnetic Resonance Imaging (MRI) Devices within FDA specified parameters
- Ob/Gyn Diagnostic Ultrasound within FDA approved parameters
Related FDA Guidance Documents: Devices
- FDA - Significant Risk and Nonsignificant Risk Medical Device Studies
- FDA - Frequently Asked Questions About Medical Devices
- FDA Decisions for Investigational Device Exemption Clinical Investigations
- FDA - Guidance on Informed Consent for In Vitro Diagnostic Device Studies Using Leftover Human Specimens that are Not Individually Identifiable
In accordance with the Federal Food, Drug, and Cosmetic Act, FDA places all medical devices into one of three regulatory classes based on the level of control necessary to ensure safety and effectiveness of the device. Classification is risk based, that is, the risk the device poses to the patient and/or the user is a major factor in determining the class to which it is assigned.
Devices in all three classes are subject to general controls which require, in part, that companies: (1) register their establishments and list the medical devices they market with FDA; (2) manufacture their devices in accordance with Good Manufacturing Practices; and (3) label their devices in accordance with labeling regulations.
Class I devices are subject only to general controls. They typically present the lowest potential for harm and are simpler in design than Class II or Class III devices. Examples of Class I devices include elastic bandages, examination gloves, and hand-held surgical instruments.
Class II devices are those for which general controls alone are insufficient to provide a reasonable assurance of safety and effectiveness. In addition to complying with general controls, Class II devices are also subject to special controls identified by the agency, which may include special labeling requirements, performance standards and postmarket surveillance. Examples of Class II devices include powered wheelchairs, infusion pumps, and surgical drapes.
Class III devices generally are those for which insufficient information exists to determine that general or special controls are sufficient to provide a reasonable assurance of safety and effectiveness. Examples of Class III devices include replacement heart valves, silicone gel-filled breast implants, and implanted cerebellar stimulators. The premarket approval process (PMA)is the FDA process of scientific and regulatory review to evaluate the safety and effectiveness of class III medical devices.
Excerpted from Information Sheet Guidance For IRBs, Clinical Investigators, and Sponsors. Frequently Asked Questions About Medical Devices, 2006
FDA Regulatory Approval Pathways
The regulatory pathway for approval by the FDA depends on whether or not there is an existing device that is substantially equivalent – a predicate device – and the number of potential patients who might use the device. Whether or not clinical trials are required to support the approval of a new device will depend on these factors.
Premarket Approval is the regulatory pathway for devices for devices that do not have a predicate device or where the new device is not substantially equivalent. These devices require reasonable evidence of safety and effectiveness to obtain FDA approval. Once a device has received FDA PMA, supplements are often submitted to modify the device. Supplements are reviewed via 5 different pathways, the majority of which do not to require any clinical trials. Manufacturers tend to release multiple minor changes rather than releasing a single substantive change allowing them to avoid the need to conduct additional clinical trials. For a more in depth discussion see Rome et al. FDA Approval of Cardiac Implantable Electronic Devices via Original and Supplement Premarket Approval Pathways, 1979-2012.
Premarket Notification - 510(k) is the regulatory pathway for devices that are substantially equivalent to a predicate device. Clinical trials and data in humans are not required for devices approved via this pathway. Requiring clinical trials for every minor change to medical devices would be expensive. Minor changes to devices as complex as heart valves, joint replacements, surgical instruments, implanted pacemakers, or insulin pumps might not require any clinical trials if there is a predicate device that can be referenced. Sometimes the ancestry of a predicate device can be quite labyrinthine and involve dozens of devices. The 510(k) Ancestry of a Metal-on-Metal Hip Implant outlines the long a twisted history of the various predicate devices that allowed metal-to-metal hip implants approval under the 510(k) pathway. For a history of the successes and failures of the 510(k) approval process see the Institute of Medicine Report Medical Devices and the Public Health. The FDA 510(k) Clearance Process at 35 Years.
Humanitarian Device Exemption is the regulatory pathway for devices for devices that are intended for certain rare conditions where it would not be possible to develop sufficient evidence of safety and effectiveness to support a PMA.
Humanitarian Device Exemption is the regulatory pathway for devices for devices that are intended for certain rare conditions where it would not be possible to develop sufficient evidence of safety and effectiveness to support a PMA.
21 CFR 812.3 Definitions
(n) HUD (humanitarian use device) means a medical device intended to benefit patients in the treatment or diagnosis of a disease or condition that affects or is manifested in fewer than 8,000 individuals in the United States per year.
(m) HDE means a premarket approval application submitted pursuant to this subpart seeking a humanitarian device exemption from the effectiveness requirements of sections 514 and 515 of the act as authorized by section 520(m)(2) of the act.
A Humanitarian Device Exemption (HDE) application is similar to a premarket approval (PMA) application. However, due to the limited size of the target population and the impracticability of conducting clincal trials to establish safety and effectiveness, an HDE is exempt from the requirement to establish reasonable assurance of effectiveness. The FDA's approval is based on its determination that the device will not expose patients to an unreasonable risk of injury or illness and the "probable benefit" to health. This is a considerably lower standard than proof of safety and effectiveness.
FDA approval of an HDE authorizes an applicant to market the HUD, subject to certain profit and use restrictions. HUDs cannot be sold for profit, except in very limited circumstances. Most importantly, they can only be used in a facility after an IRB has approved their use in that facility, except in certain emergencies.
How is an HDE different from an IDE?
An IDE is an exemption that allows a sponsor to ship an investigational device prior to marketing approval; it authorizes the investigational use of the device. A HUD with an approved HDE is approved for marketing. This means that the sponsor can ship the device and can bill for its use (but cannot make a profit from its sale except under limited circumstances). To receive approval for an HDE, the FDA must conclude that there is no comparable device available to treat the disease or condition. If a comparable device is approved by the FDA, the HDE approval may be withdrawn. An HDE therefore is analogous to a PMA application; its an application to market the device. An IDE is an application to ship an investigational device to conduct studies.
To approve a PMA device, the sponsor has to provide data from adequate and well-controlled trials that provide substantial evidence of effectiveness (and safety). The requirements for approval of an HDE application to market a HUD are quite a bit lower. FDA's Q&A states that an approved HDE represents "a determination by FDA that the HUD will not expose patients to an unreasonable or significant risk of illness or injury and the probable benefit to health from use of the device outweighs the risk of injury or illness from its use while taking into account the probable risks and benefits of currently available devices or alternative forms of treatment."
What are the IRB's Responsibilities for a HUD?
A. Clinical use of a HUD for treatment or diagnosis in accordance with the approved label or off-label
-
Clinical use of a HUD for treatment or diagnosis in accordance with the approved label or off-label
After HDE approval, a HUD may only be used after IRB approval has been obtained for the use of the device, even for the FDA-approved indication, except for an emergency use situation. The IRB is required to perform its initial review at a convened meeting using the criteria for approval at 21 CFR 56.111. Subsequent continuing reviews - at least annually - may be performed using expedited procedures. Expedited review is permitted because a HUD is an approved device.
-
Emergency use of a HUD for clinical care in a single patient
The healthcare provider is required to comply with the requirements of the Emergency Use of Investigational Drugs, Biologics, or Devices SOP (IRB SOP 802, V.C.1-3, and 5). The IRB will review the 5-day report in accordance with IRB SOP 802.
B. Investigational use of a HUD in accordance with the approved label or off-label
-
Investigational use in accordance with the approved label to collect safety and effectiveness data
The IRB will review the initial study at the convened board.
-
Investigational use off label
The IRB will review the study at the convened board. The proposed use will need to be in compliance with FDA regulations under 21 CFR 50, 56, and 812 (e.g. may require submission of an IDE application to FDA or be conducted under the abbreviated requirements for NSR devices at 21 CFR 812.2(b)).
For details on the requirements for initial submission to the IRB, continuing reviews, amendments, and adverse event reporting, see the Humanitarian Use Device (HUD) SOP (IRB SOP 412).
References
- 21 CFR 814. See Subpart H - Humanitarian Use Devices, which contain the regulations governing HUDs.
- FDA Guidance Document Humanitarian Use Device (HUD) Designations issued September 5, 2019.
- FDA Guidance Document Humanitarian Device Exemption (HDE) Program issued September 6, 2019.
- Institutional Review Boards and Humanitarian Use Device (HUD) Powerpoint learning module presented by Dr. Fabienne Santel of the Center for Devices and Radiological Health at the FDA.
Frequently Asked Questions
-
What types of devices need to be included in the eIRB application?
(1) Unapproved devices, (2) all investigational devices, meaning all those being studied for their safety or effectiveness regardless of whether they are FDA-approved/cleared or not, and (3) FDA-approved/cleared devices that are not approved for use for clinical use at CHOP.
-
How do I include a device in the eIRB application?
Check the "Device" box in Section 5.04 (2.0). This will then require completion of Section 8.02. The information about the device - e.g. brochures, manuals, 510(K) clearance approval letters - should all be attached in Section 12.02 (3.0). Risk information should be included in the protocol, consent form and in eIRB Section 11.01 (1.0).
-
The device is not a physical instrument, it's medical software, a diagnostic test/assay, an MRI sequence, or a medical app. What type of storage plan is required?
There needs to be a plan that explains how the use of the device is only used for this research study and that will limit access to members of the investigative team. For example, there needs to be a plan to limit the use of unapproved MRI sequences so that they are not used for clinical care.
-
Who decides whether a device is non-significant risk or significant risk?
The sponsor is responsible for making the initial NSR-SR determination. The IRB is responsible for reviewing and concurring with or disagreeing with, the sponsor's assessment. If the sponsor has a letter from the FDA indicating that the device is NSR, the IRB must accept that decision (the FDA's determination is final). Otherwise, if the IRB disagrees with the sponsor's determination that the device is NSR, the sponsor must request that the FDA issue its opinion.
-
If the IRB has determined that the device meets the definition of an NSR device, do I need to get an IDE from the FDA?
No. If the IRB concurs with the sponsor's NSR determination, then it is effectively issuing an abbreviated IDE to conduct the study. NSR device studies must follow the abbreviated requirements under 21 CFR 812.2(b) and the FDA's regulations for Human Subjects Protections and the IRB operations (21 CFR 50 and 56).
-
My sponsor initially determined my device is NSR but the IRB determined it was SR. What do I need to do?
The sponsor must obtain one of the following from the FDA: (1) a determination that the device is an NSR device or (2) an IDE (in the event the FDA agrees that the device is an SR device).
-
My sponsor initially determined the device is an SR device. Can I submit the study to the IRB for review if the FDA hasn't issued the IDE yet?
Yes. However, the IRB will not release its final approval until an IDE has been issued and documentation, including the IDE number, has been provided to the IRB.