HOW CAN WE HELP YOU? Call 1-800-TRY-CHOP

In This Section
Could Improved Mitochondrial Function Affect Pediatric Neurodegeneration?
Published on October 21, 2021 in Cornerstone Blog · Last updated 2 months 3 weeks ago
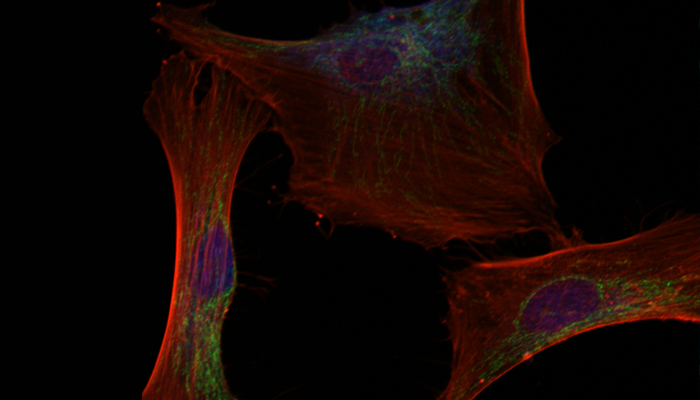
TBCK-patient derived fibroblasts generated in the Ortiz-Gonzalez lab.
The findings:
Children's Hospital of Philadelphia researchers examined fibroblasts from patients with biallelic mutations of the TBCK gene and found that while mitochondria had no intrinsic defects, cells of patients with the TBCK gene mutation have a deficit in the mitophagy process, which typically enables the degradation and recycling of cellular components. When this lysosome-dependent function is disrupted, "old" mitochondria that would usually be flushed from the body accumulate.
Their data demonstrated impaired mitochondrial quality control, and the degree of mitochondrial dysfunction correlated with neurodegenerative course in TBCK syndrome, an ultra-rare genetic disorder that affects approximately 75 to 100 children worldwide who experience a wide-range of symptoms such as global developmental delays, congenital hypotonia, hypothyroidism, respiratory insufficiency, and dysphagia.
In addition, the researchers observed that by acidifying lysosomes using nanoparticles, the mitochondrial respiratory defects in fibroblasts were repaired, suggesting that the mutant TBCK gene impaired mitochondrial quality control secondary to lysosomal dysfunction. Their findings suggest lysosomal acidification as a potential treatment strategy to be tested in further studies.
Why it matters:
Among children with TBCK syndrome, there are differences in severity, progression, and degree of neurologic symptoms, which begs the question of why. Patients with severe disease have evidence of mitochondrial dysfunction, leading the research team to ask whether mitochondria are the culprit or the mediator. Some children who have the TBCK gene mutation experience static encephalopathy, which manifests as developmental, motor, and language delays. These patients progress along their own developmental curve, building skills from their unique baseline. Others, particularly those with the Boricua mutation, start out at a similar baseline but then decline. The study's scientific relevance is that it challenges the notion that all children diagnosed with the same rare genetic disease will experience a similar disease course, according to Xilma Ortiz-Gonzalez, MD, PhD, corresponding author.
Additionally, the findings provide insight into the disease mechanisms of TBCK encephaloneuronopathy and reveal the potential role of mitochondrial function as a biomarker beyond primary mitochondrial disorders. The study also supports further investigation of lysosomal acidification strategies for disorders of impaired lysosomal degradation affecting mitochondrial quality control.
Who conducted the study:
Lead author Jesus Tintos-Hernandez, PhD, division of Neurology, published the paper along with co-authors Kierstin Keller, BS, CGC, department of Genetics, Adrian Santana, BA, division of Neurology, all with the CHOP Center for Mitochondrial and Epigenetic Medicine, and Dr. Ortiz-Gonzalez, a physician-scientist in the department of Neurology and the Epilepsy Neurogenetics Initiative in the Perelman School of Medicine at the University of Pennsylvania.
How they did it:
The study team tested fibroblasts from patients with TBCK syndrome with various levels of clinical severity to determine a potential biomarker of neurodegenerative progression. They examined the degree of mitophagy, the natural process of selectively removing "old" mitochondria, essentially the "incinerator" for cells the body no longer needs, to determine where in the process quality control was failing. Since mitophagy is dependent on lysosomal degradation, the team examined lysosomal function and found that lysosomal proteolytic function is significantly reduced in TBCK fibroblasts.
The investigators then observed that targeting the lysosomes and decreasing the pH seemed to fix the mitochondrial problems, demonstrating that the autophagy cascade and lysosomal function had, indeed, been disrupted. When the mitochondria are efficiently maintained, degraded, and recycled, the mitochondrial function improved.
Quick thoughts:
"We took these cells from patients who had a milder course and a severe course and investigated if the mitochondrial function could serve as a marker of the severity of the disease" Dr. Ortiz-Gonzalez said. "We found that the mitochondrial dysfunction was significantly decreased in the patients with a progressive degenerative course."
What's next?
Dr. Ortiz-Gonzalez and her team will continue to explore the autophagy cascade and will complete a proof-of-concept study with lysosomal acidification in human neurons. The researchers aim to show that decreasing the pH in lysosomes using targeted nanoparticles restores normal mitophagy, with the goal of identifying a druggable target.
While the small number of patients with TBCK syndrome may prove challenging for anticipated future clinical trials, Dr. Ortiz-Gonzalez anticipates existing partnerships with patient advocacy organizations such as the TBCK Foundation will help scientists reach all potential participants.
Opportunities also exist for application with other disorders that affect autophagy, such as Beta-propeller protein-associated neurodegeneration (BPAN) and Vici syndrome. Studying these mechanistically related, genetically defined disorders could lead to common therapeutic targets.
Where the study was published:
The paper appears in Brain Communications.
Read more in the CHOP press release.
You may also like

Learn how the NAD metabolite controls how well T and B cells can detect their antigens, leading to quicker immune responses.

With the help of an artificial intelligence deep-learning algorithm called CytoCommunity, researchers better understand how specific cell groups in tissues organize to support tissue function.

CHOP researchers test novel system to help patients with severe complication related to congenital heart disease avoid open heart surgery.